We have covered a lot of ground in this Automating & Standardizing Your Workflows blog series. First, we saw how to perform secondary analysis with Sentieon to generate the necessary VCF and BAM files for tertiary analysis in Part I. The implementation of VSPipeline allowed for rapid import and project generation for a predefined cancer gene panel project template in VarSeq covered in Part II. Then, we moved into an in-depth tertiary analysis and variant interpretation via VSClinical in Part III.
Now, the next subject to cover is how to leverage a genomic repository which stores the massive amount of NGS data. Storage alone is critical, but how else can a user benefit from querying through stored data, maintain awareness of evolving clinical evidence for all cohort data, and then make this repository available for multiple users all performing analysis? The solution to this complex problem is VSWarehouse.
Moreover, VSWarehouse can also be connected to your pre-existing LIMS (Lab Information Manage System) for an even more robust data storage solution. VSWarehouse is accessed in two ways: from a terminal in the VarSeq project and from a browser interface setup to query through all stored content. Let me break these down individually.
VSWarehouse + VarSeq terminal
VarSeq contains a specific “VConnect” icon which can be used to link to the installed VSWarehouse instance on a designated server (Figure 1, below). From the “VConnect” terminal, users can upload new samples from their pipeline into existing projects contained on the VSWarehouse server. This is also the access point to create VSWarehouse based assessment catalogs and clinical reports.
Regarding samples and projects, the value in storing data in VSWarehouse could be to annotate against panel cohorts to eliminate common variants or false-positive artifacts. As users process more samples over time, these samples can be continuously added to the cohort to add more power to this filtering of common or artifact variants. Producing VSWarehouse based catalogs and reports is crucial to clinical consistency and efficiency by creating comprehensive classification catalogs all users can submit to and standardized reports customized to each panel being run.
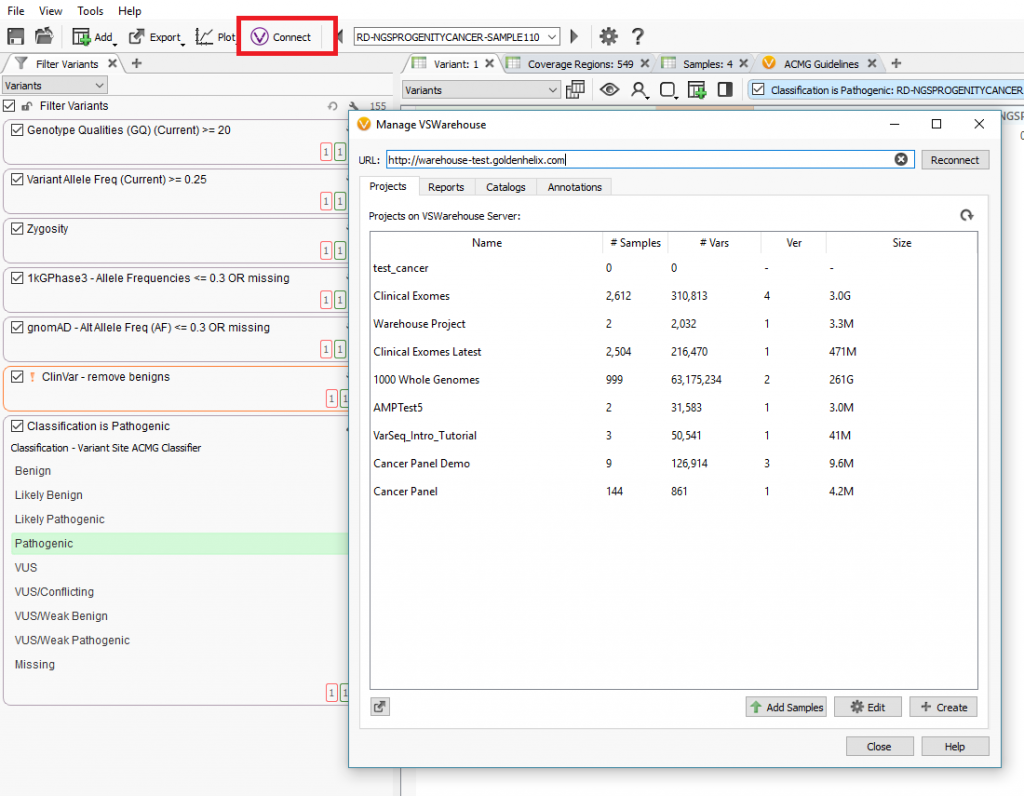
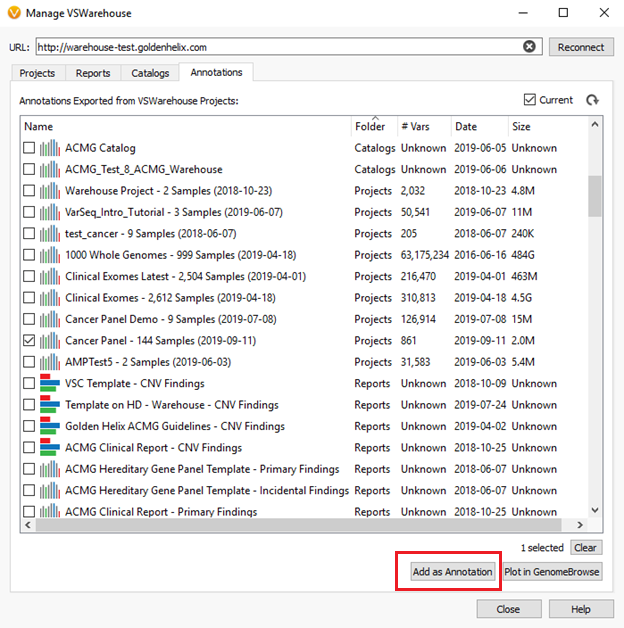
To annotate against cohort data, users can switch over to the “Annotations” tab in the VSWarehouse terminal. Figure 2, below, shows an example of this where the Cancer Panel project containing 144 samples could be used as a variant allele frequency annotation in the current VarSeq project being run.
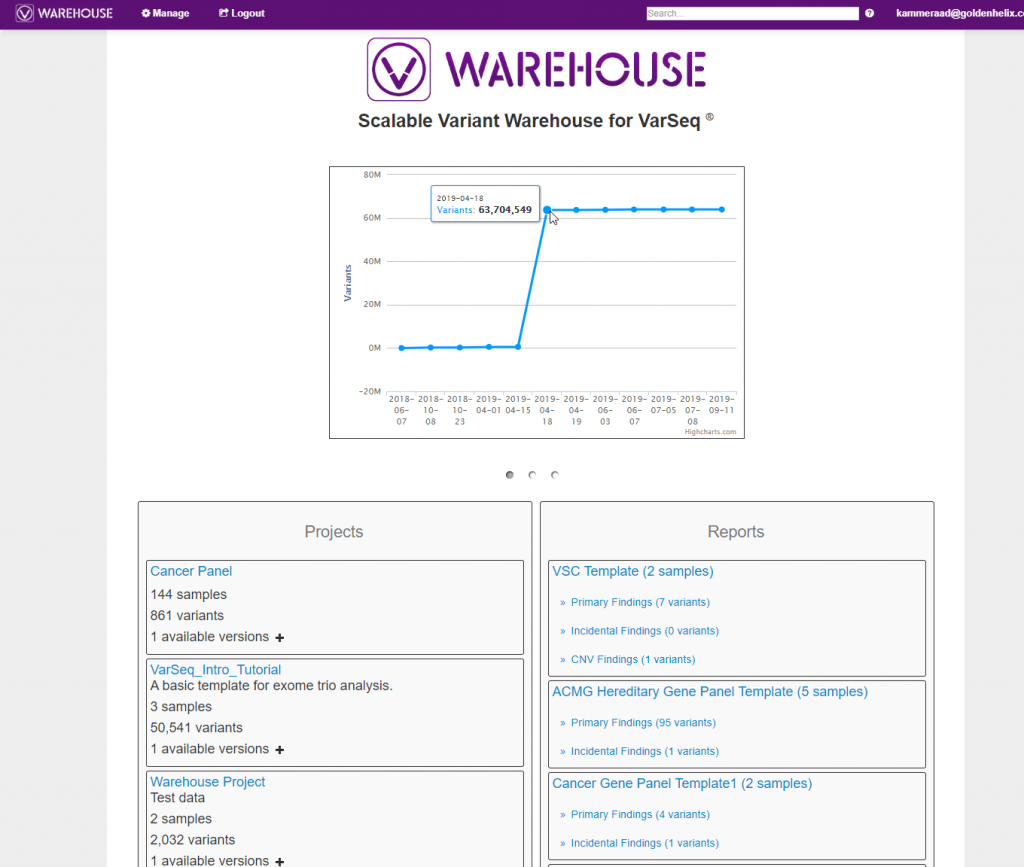
As mentioned previously, the stored content can also be queried through the VSWarehouse browser. In Figure 3, below, you can see a snapshot of the VSWarehouse home page listing the total number variants stored (over 63 million in this example!), along with a list of stored projects, reports, and catalogs.
Querying Through Stored Content
The querying power within VSClinical is shown in Figure 4, below, where a filter logic was built to isolate known pathogenic variants from ClinVar, among all variants in the cancer panel cohort.
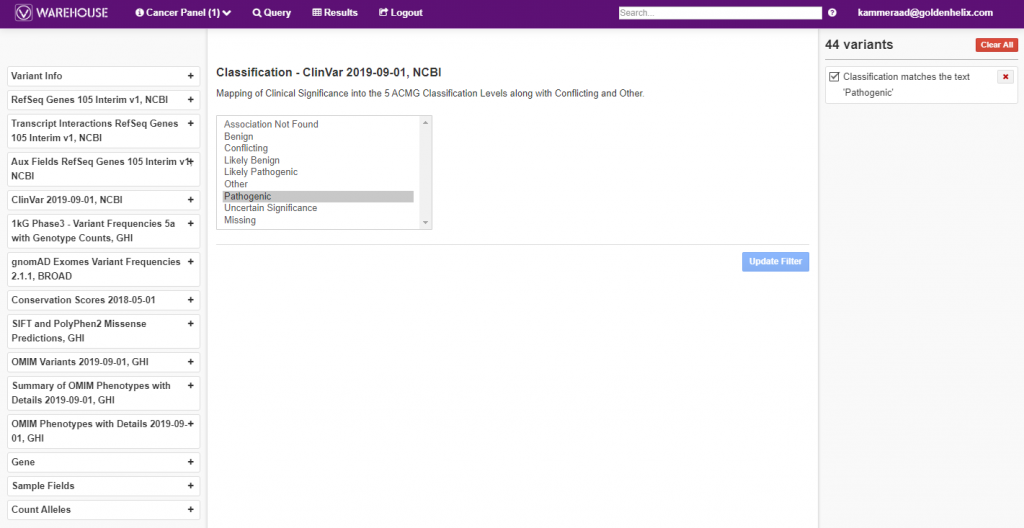
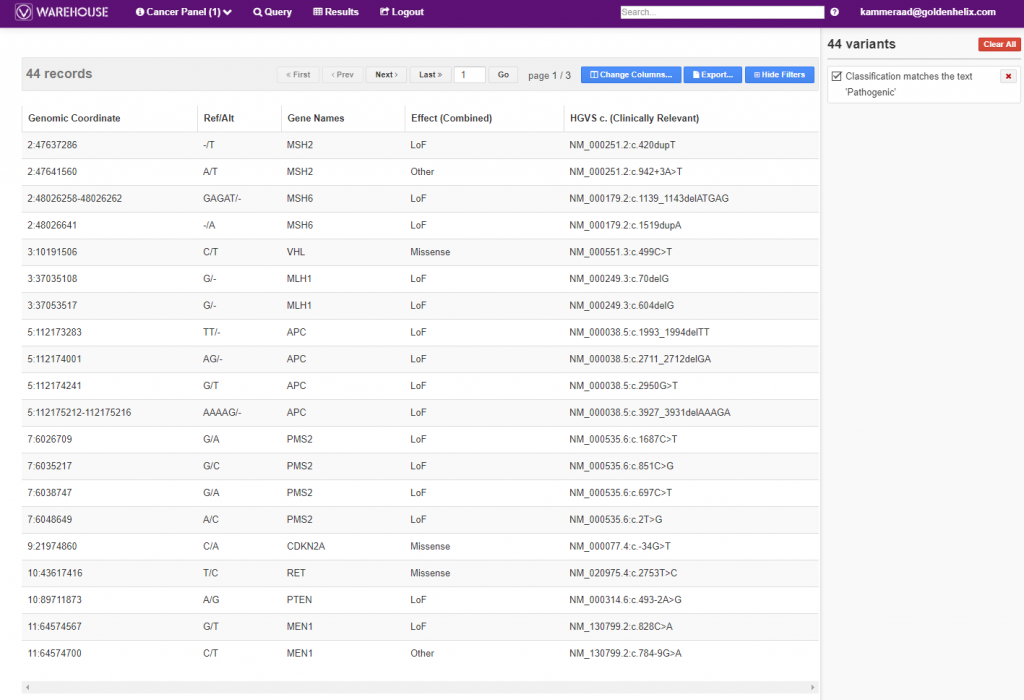
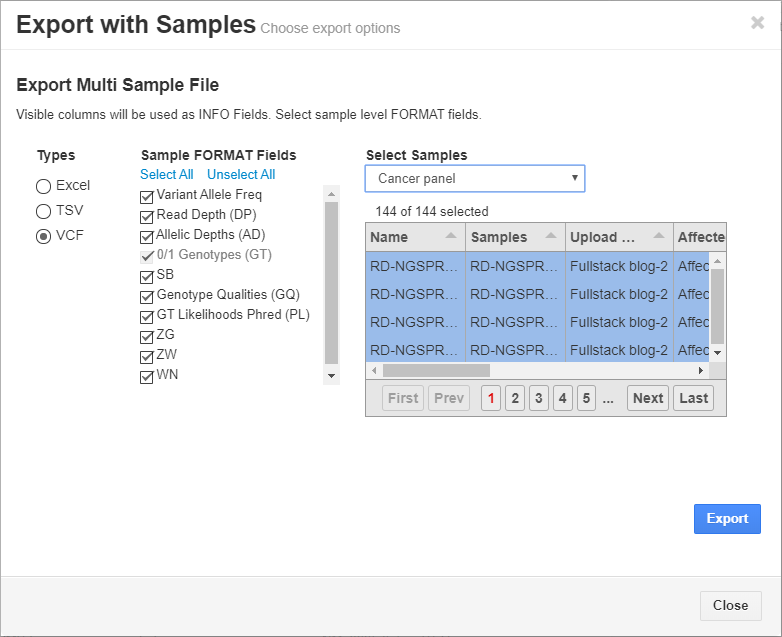
Additionally, the final, filtered results show 44 variants (Figure 5, below), all of which can be exported out with cohort sample sets into VCF or additional formats (Figure 6, below).

Reviewing New and Changing Variants from ClinVar
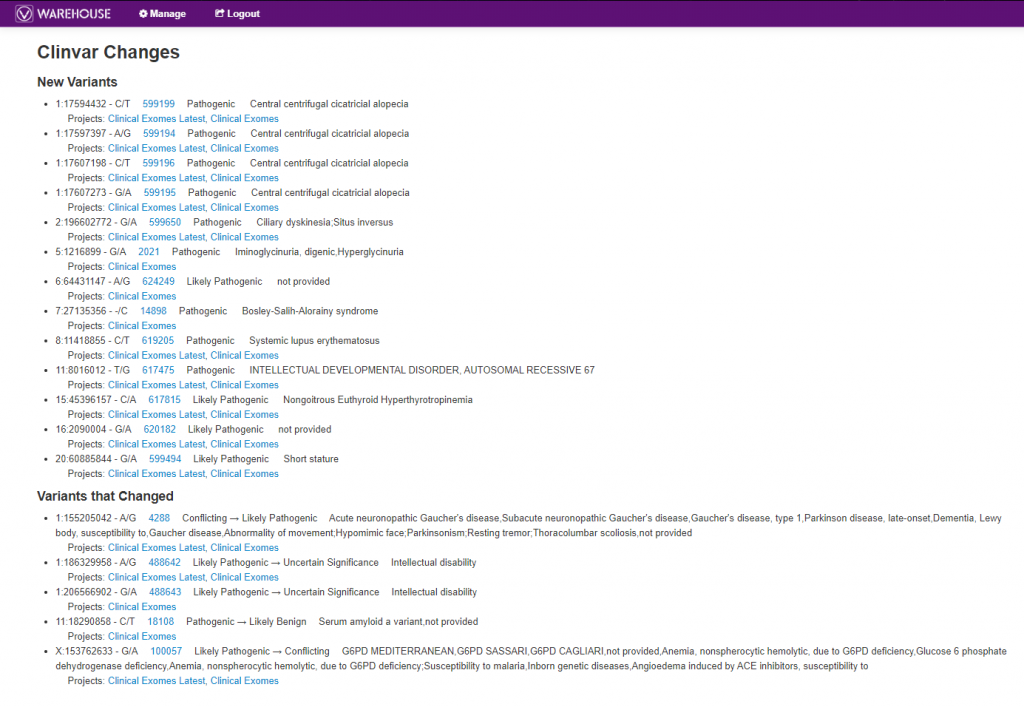
Another extremely powerful tool worth mentioning in VSWarehouse is accessing the changing classification knowledge from databases like ClinVar. As seen in Figure 7, below, users can get a list of all new variants submitted to ClinVar with their related classifications, as well as see changing classifications much like the top listed “Variants that Changed” with the “Conflicting -> Likely Pathogenic” update. Both of these scenarios are crucial in following up with a patient where the variant may have had an overall stale or uneventful interpretation but now can be re-evaluated. The links to projects each of these variants are present in are also included, making the follow-up process even more efficient.
Managing User Access
Any admin user can manage user access to projects, catalogs, and reports by clicking on the “Manage” gear-shaped icon (Figure 8, below). VSWarehouse allows full control of accessibility even down to individual fields in the project. Even collaborators simply accessing VSWarehouse can review the contents of project results but be prevented from accessing or modifying the projects themselves.

I hope you enjoyed this full-stack blog series and that it provided a bit more clarity our software solutions. It is our goal to assist in the standardization of your workflows and create the most efficient, user-friendly experience without losing any of the crucial analysis or data management issues you may face. Please contact info@goldenhelix.com if you wish to know more details about the subjects covered in this blog series.