In our last part of this series, we showed how to run a pre-built workflow template via VSPipeline to automatically import and filter sample variants to streamline the search for clinically relevant variants. Now, we can deep-dive into our filtered, pathogenic variants to fully understand and capture their final classification and interpretation.
Filtered Germline Variants for ACMG Guidelines
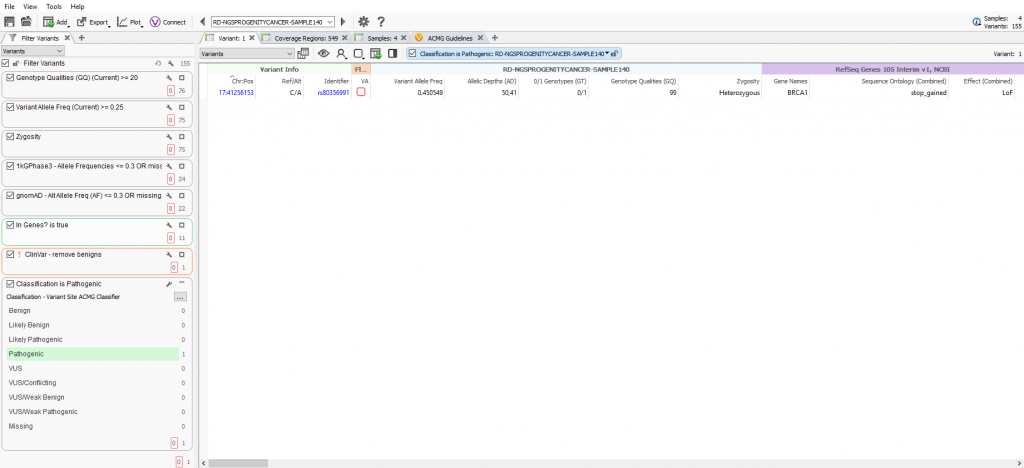
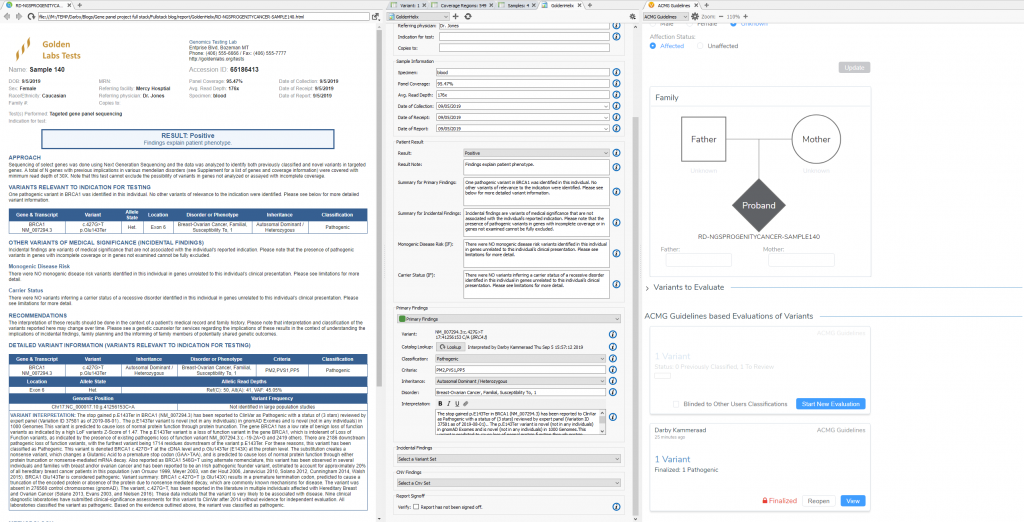
The VSPipeline output in back in Part II generated a final project of filtered variants for four samples in a cancer gene panel project template. The final list of filtered variants are all recommended as pathogenic from the ACMG autoclassifier algorithm integrated into the VarSeq project template. These “pathogenic” variants can now be flagged for evaluation in the VSClinical to confirm the recommendation (Figure 1).
Variant Interpretation and Classification via VSClinical
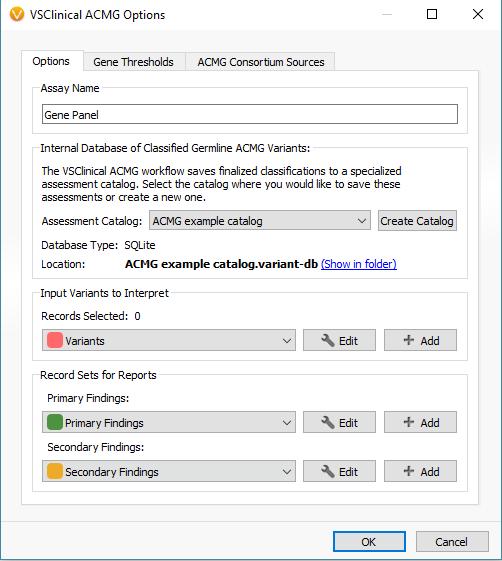
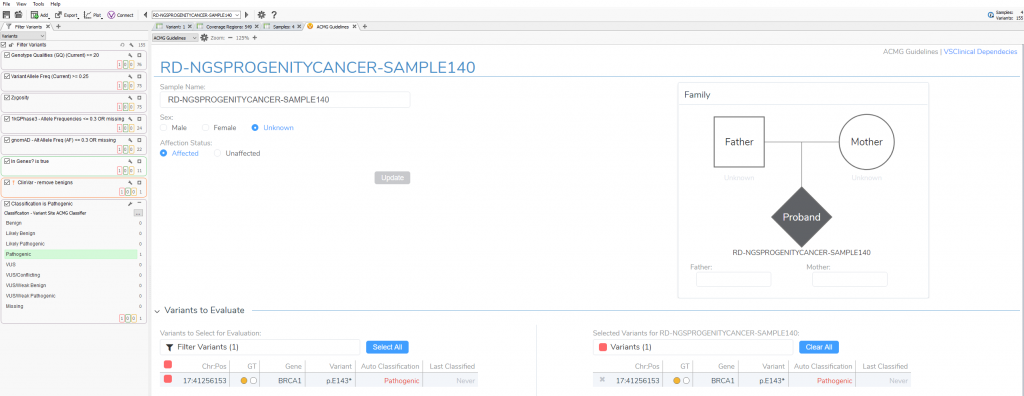
When first opening VSClinical, you will be prompted with the “Options” menu to select the assessment catalog, variant sets for evaluation, and primary/secondary findings (Figure 2). This is also where you can modify allele frequency thresholds in the “Gene Thresholds” tab. Once the catalog and options are in place, the evaluation can begin with the filtered variant selected (Figure 3).
VSClinical Step 1: Classification Tab
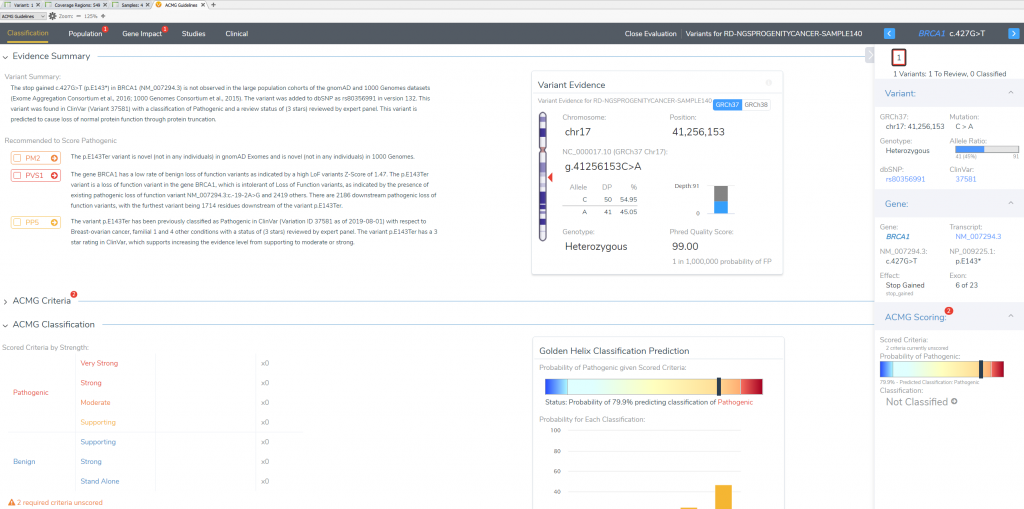
This variant’s interpretation begins at the “Classification Tab” which summarizes the effect of the variant and lists the recommended criteria left to evaluate. This will also be the final tab to define the ultimate classification and interpretation for this BRCA1 stop gain variant in exon 6 of 23.
VSClinical Step 2: Population Tab
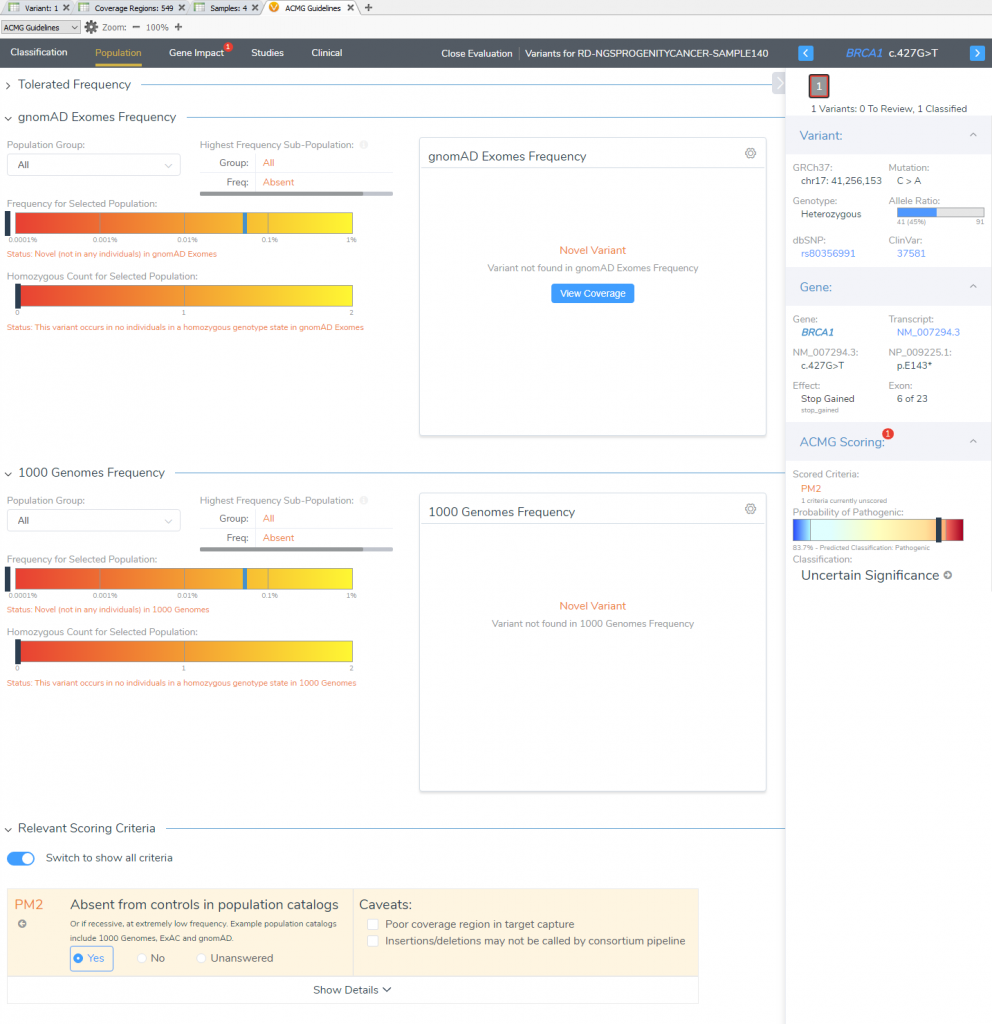
The “Population” section quickly notifies the user that this particular variant is novel for both frequency catalogs (1kGPhase 3 and gnomAD). This allows for the capture of our first moderate pathogenic criteria (PM2).
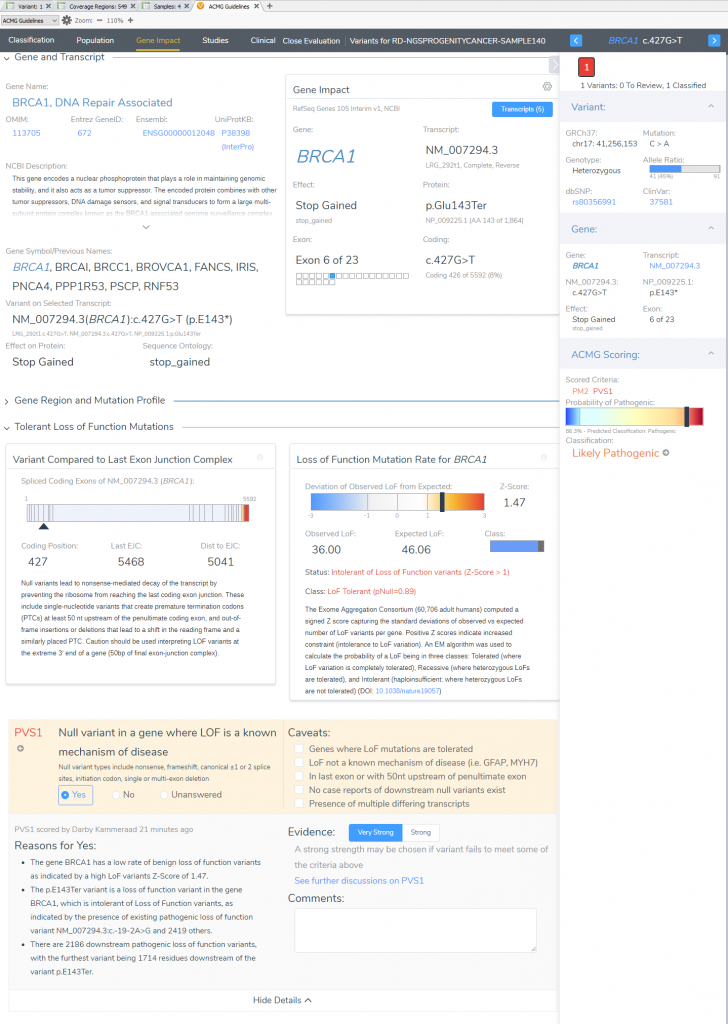
VSClinical Step 3: Gene Impact
In the “Gene Impact” section, we see that this variant is one among many other pathogenic LoF variants in BRCA1, which allows for the inclusion of the strongest pathogenic criteria (PVS1: Null variant in gene where LoF is a known mechanism of disease). The finer details here report that BRCA1 has a low rate of benign LoF variants with a high Z-Score of 1.47, other pathogenic LoF variants are known in BRCA1, and 2186 of those known LoF variants exist downstream from this one. You may notice that the summation of PM2 and PVS1 has brought us to likely pathogenic; to reach final pathogenicity as recommended requires literature review of similar known pathogenic variants.
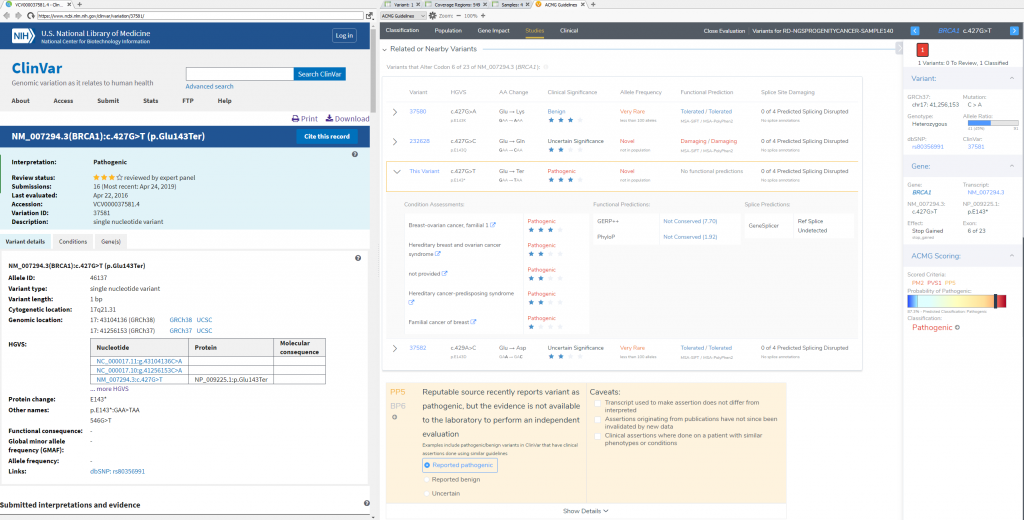
VSClinical Step 4: Studies
To confirm the pathogenicity of any variant, it is always critical to review the available literature and submissions of previously reported variants in a database like ClinVar. VSClinical provides a simple approach to not only direct link to a variants’ submission in ClinVar, but can also easily search across PubMed and Google to find additional content. This specific variant has 16 ClinVar assessments available, and a multiple star status for the matching condition for this particular patient. For this stop gain, the recommendation is to include PP5 supporting criteria (Reputable source reports variant as pathogenic) which gets the final classification of the variant to Pathogenic. The studies section will also allow for direct capture of clinical text from these variant submissions, and citation of the PubMed IDs for final interpretation.

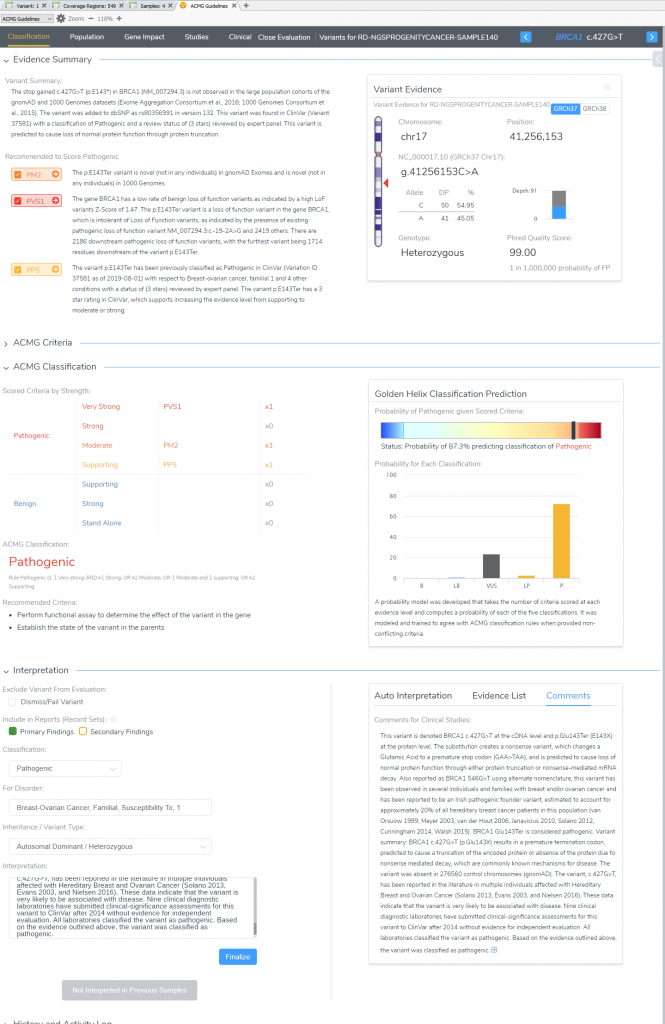
VSClinical Step 5: Variant Classification and Interpretation Capture for Report
Now that all relevant criteria have been reviewed and literature assessed, the last step in VSClinical is to store the classification and interpretation for this variant. All selected criteria will populate the “ACMG Classification” table, with an additional listing of the ACMG rule to classification. This is convenient for future reference in seeing this variant in future samples, and also streamlines the rendering of clinical reports. Once we review the content and finalize this sample, we can then easily produce the report all from VarSeq (Figure 9).
Now we are ready to explore how to capture and store this project data into VSWarehouse, our genomic repository which is covered in our next part of this blog series. The exploration of VSWarehouse will cover how to upload new sample and variant data into the repository, and how to leverage the stored data against future projects. The value points here include isolating rare or novel variants not seen among your private cohort, simple removal of false-positive variants or bioinformatic artifacts, and simple ways to remain up-to-date on ever-evolving variant classifications and evidence for your cohort variants over time. Don’t forget you can always reach out to support@goldenhelix.com if you have any questions regarding ACMG guideline utilization in VSClinical or any other feature discussed in this blog series.