Overview
VSClinical enables users to evaluate variants according to the ACMG guidelines in a high-throughput fashion and obtain consistent results and accurate variant interpretations. This feature is tightly integrated into our VarSeq platform as well, and when paired together, users can evaluate NGS data and obtain clinical reports all in one suite. Coupled with the ability to find novel or rare variants, this suite can be used to identify variants associated with specific disorders. In this blog, we will start with a hypothetical case study with a known disease phenotype and identify and evaluate the causal variant according to the ACMG guidelines.
Case Study
A 30-year-old male patient displayed symptoms of dehydration and disorientation as well as progressive stiffness in the legs (paraparesis) and excessive muscle tone (hypertonia). Blood analysis was requested and revealed an increase in very long chain fatty acids (VLCFA), which raised a clinical suspicion of adrenal insufficiency and possibly adrenoleukodystrophy (ALD). ALD is a disorder of peroxisomal fatty acid beta-oxidation, which results in the accumulation of VLCFA, which can deteriorate the myelin tissue in the central nervous system (1). Since ALD is a genetic disorder, NGS sequencing was requested, and the output was processed through VarSeq for filtering of the variants and VSClinical was implemented to evaluate the candidate variant(s) according to the ACMG guidelines.
Clinical Assessment
As mentioned in Part I of this blog series, the PhoRank algorithm can be used for prioritizing variants for targeted disorders. Alternatively, a similar algorithm for this prioritization is the Match Genes Linked to Phenotypes. This algorithm is helpful when wanting to avoid the “rank” specificity as output in PhoRank, and instead search across the associated genes in their entirety. For this case study, Match Genes Linked to Phenotype was implemented with the field name Motor Sensory Loss and utilized the ALD associated phenotypes the patient was displaying: paraparesis, hypertonia and adrenal insufficiency (Figure 1). Entering in the phenotypes into this algorithm generated a list of genes and identified a single G>A missense variant in the ABCD1 gene of the sample.
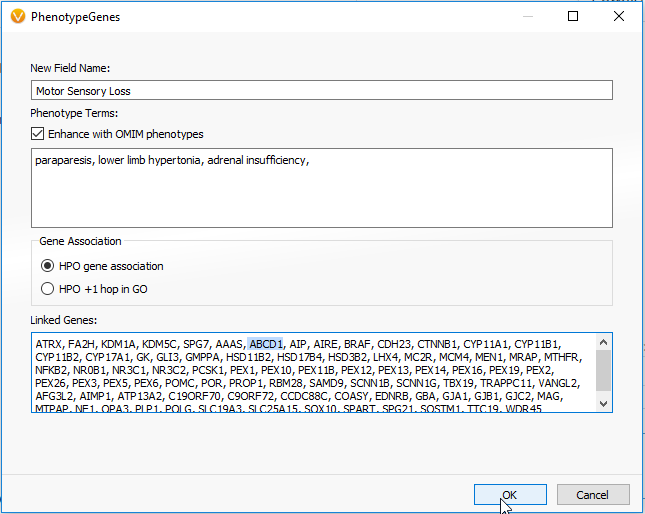
Frequency of the Variant c.1661G>A in the ABCD1 gene
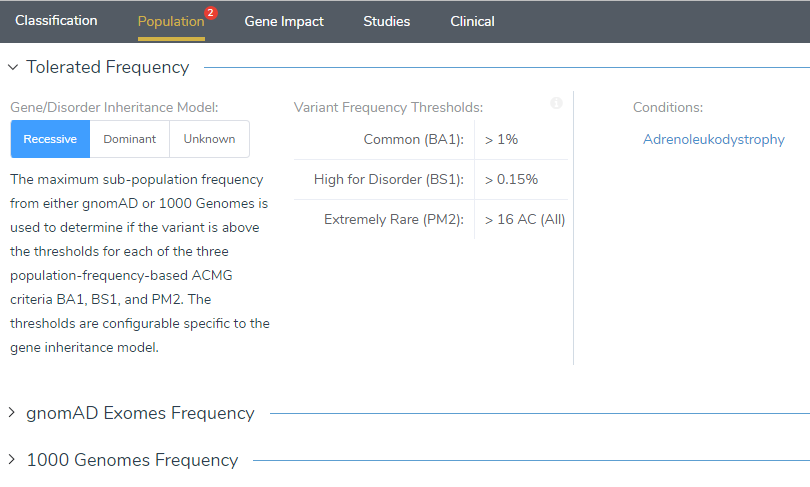
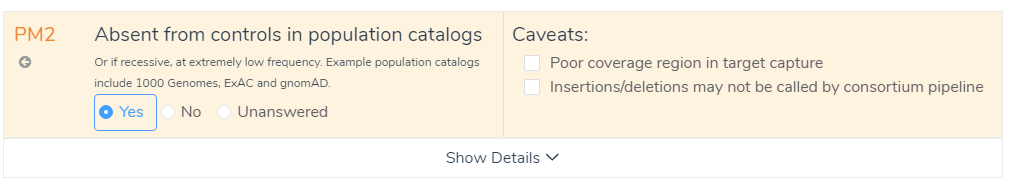
The next step in the evaluation process is scoring and interpreting the variant in VSClinical. To begin, we will focus on the frequency of the variant in the Population tab. In this tab, we can first look at the Tolerated Frequency, which determines the threshold used to determine if a variant is rare or novel among sub-populations in gnomAD Exomes or 1000 Genomes (Figure 2). The missense variant c.1661G>A in the ABCD1 gene has been characterized as causing an X-linked recessive genetic disorder called adrenoleukodystrophy (2). This means that males with this mutation will develop the disease as they only have one X-chromosome, while females with this mutation will be carriers as they have two X-chromosomes. As shown in Figure 2, this variant has been identified as having a recessive disorder inheritance model, which provided the designated frequency thresholds (Figure 2). With these thresholds applied, this variant was found to be novel in gnomAD Exomes and in 1000 Genomes Frequency, which led to answering Yes to the ACMG criteria PM2.
Variant Gene Impact
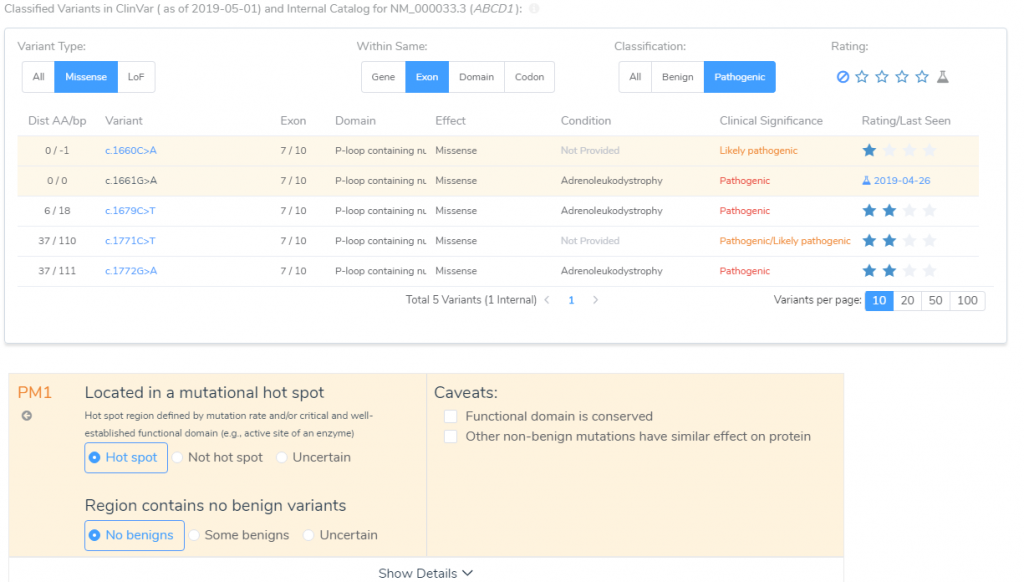
Next to investigate in VSClinical is the Gene Impact tab. According to the Gene Region and Mutation profile, there have been 5 previous classifications of neighboring missense variants that occur in the same exon with classifications of Likely Pathogenic or Pathogenic with their associated review status (Figure 3). With this evidence, PM1 was answered for this variant, which is that this variant is located in a mutational hotspot and there are no benign variants located in the region.
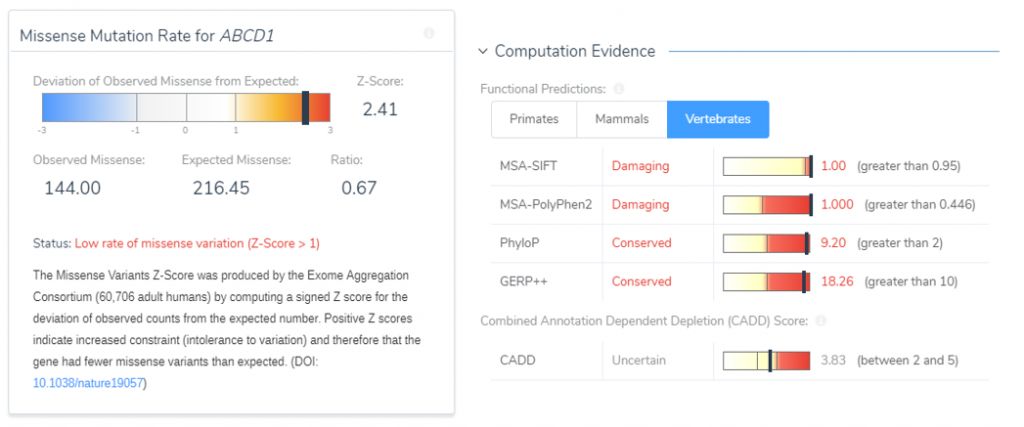
The Gene Impact tab also displays the genes intolerance to the missense variant based on a computed Z-score and will show the influence of the variant on functional prediction algorithms. This variant’s Z-score was 2.41, indicating that this gene has a low rate of benign missense variation (Figure 4). Using the available variant evidence, VSClinical also identified that the ABCD1 gene contains 49 pathogenic missense variants, indicating that missense variants are a common mechanism of disease. This information provided strong evidence for answering PP2 criteria: this gene has a low rate of benign missense variation and missense variants in this gene are causal. Furthermore, this variant was predicted to have a damaging effect according to SIFT and PolyPhen and occurred in a conserved region according to PhyloP and GERP++. This evidence was used to answer PP3, which states that multiple lines of computational evidence support a deleterious effect on the gene or gene product.
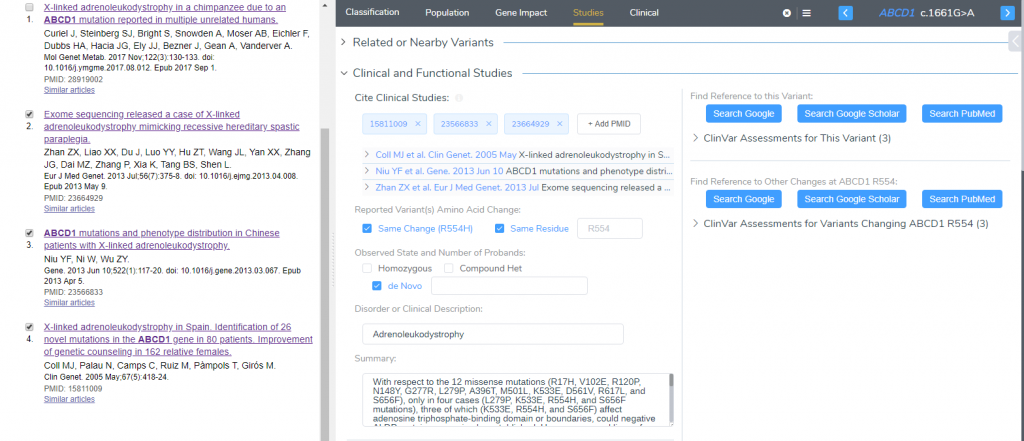
Clinical Studies
After looking at the population frequency and the influence of the variant on the gene, we can conclude the evaluation by assessing if there is any available literature in the Studies tab. In this section, we display all submissions according to ClinVar Assessments and provide the user the ability to search Google, Google Scholar and for articles in PubMed. For this variant, four publications resulted from searching PubMed, three of which were used as they had documentation of the same variant causing X-linked adrenoleukodystrophy (Figure 5). These publications were cited in the Clinical Studies for having the same amino acid change and residue as the current variant. Using this evidence allowed both PS1 and PM5 to scored.
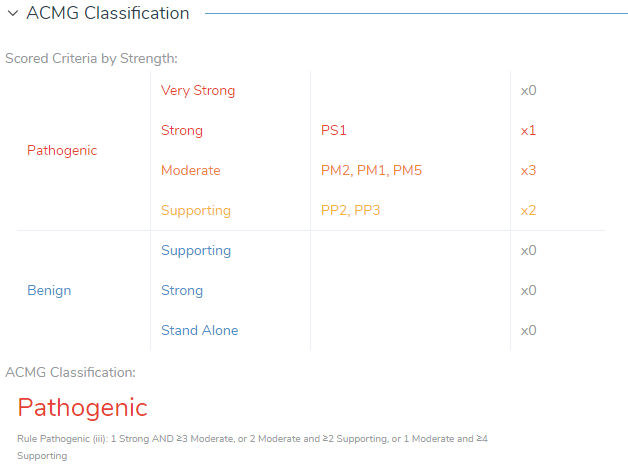
Clinical Variant Summary
In summary, VSClinical allows users to deep dive into the variant evidence and available literature to evaluate and interpret variants according to the ACMG guidelines. Using resources within VSClinical, we identified that X-linked adrenoleukodystrophy (X-ALD) is a genetic disorder that results in deterioration of the myelin sheath that insulates the nerves in the brain (3). This deterioration is a result of a deficiency in the adrenoleukodystrophy protein (ALDP), which can occur as a result of mutations in the ABCD1 gene (3). The candidate variant in the ABCD1 gene was identified using the Match Genes Linked to Phenotype using the associated signs and symptoms of paraplegia, hypertonia, and adrenal insufficiency. In VSClinical, the variant was characterized with a recessive genetic disorder inheritance model and found to be novel in 1000 Genomes and gnomAD Exomes. ABCD1 was shown to have a low rate of benign missense variation and contained 49 other pathogenic missense variants, indicating that missense variants are a common mechanism of disease. VSClinical also identified that this variant is predicted to be damaging by both SIFT and PolyPhen2 and predicted conserved by GERP++ and PhyloP across 100 vertebrates. Together, with cited clinical studies, the final classification of the variant was found to be Pathogenic according to the ACMG rule logic iii, which was answered using 1 Strong (PS1) and 3 Moderate criteria (PM2, PM1, PM5), as well as 2 Supporting criteria (PP2, PP3) (Figure 6).
Hopefully, this was an insightful read and gave you details into how you can leverage VarSeq and VSClinical to evaluate variants from NGS data that correlate to a known phenotype. Additionally, this blog was meant to showcase the software’s capabilities and does not constitute a final clinical assessment from healthcare professionals. If you have any questions or comments, please leave them in the comment section or contact us at support@goldenhelix.com.

Check out some of the other blogs in this series:
- Variant Interpretation with VSClinical: Clinical Example for Congenital Indifference to Pain
- Variant Interpretation with VSClinical: Evaluation of Hypertrophic Cardiomyopathy
- Variant Interpretation with VSClinical: Congenital Myasthenic Syndromes (CMS)
- Variant Interpretation with VSClinical: Huntington’s Disease (HD)
- Variant Interpretation with VSClinical: Non-Small Cell Lung Cancer
- Variant Interpretation with VSClinical: RET-KIF5b Gene Fusion
Citations:
- Inderjit Singh and Aurora Pujol. Pathomechanisms Underlying X-Adrenoleukodystrophy: A Three-Hit Hypothesis. Brain Pathology. 2010 Jul; 20(4): 838-844.
- Zi-xiong Zhan et al. Exome Sequencing release a case of X-linked adrenoleukodystrophy mimicking recessive hereditary spastic paraplegia. European Journal of Medical Genetics. 2013; 56: 375-378.
- Kirby Smih et al. X-Linked Adrenoleukodystrophy: Genes, Mutations, and Phenotypes. Nuerochemical research. 1999; 24(4):521-523.