Congenital Myasthenic Syndromes (CMS) History:
Congenital Myasthenic Syndromes (CMS) are a group of rare hereditary conditions that can cause seizures, severe muscle weakness, respiratory problems, and potentially disabling weaknesses shortly after birth or early childhood (1). CMS is the result of abnormalities in acetylcholine proteins residing in the motor endplate of the neuromuscular junction (1). These abnormalities can be diagnosed using magnetic resonance imaging (MRI) and electromyography (EMG) and tested using a gene panel assay for mutations in CMS-associated genes including COLQ, CHAT, CHRNA1, CHRNB1, CHRND, CHRNE, RAPSN, MUSK, DOK7, and SCN41 (2). Understanding the specific mutation(s) causing CMS is also important for determining treatment, as certain mutations lead to treatment-responsive conditions while others do not.
Case Report
The patient was born full term following a normal delivery and had a generalized seizure on day 7 and again at age 2 months. The patient displayed ptosis (droopy eyelids) and blood tests were negative for Ach receptor antibodies in the serum. Electrodiagnostic testing was requested and showed a decremental EMG response of the compound muscle action potential (CMAP). Based on these findings the patient was diagnosed with CMS and genetic testing was requested to determine available treatments.
Genetic Testing
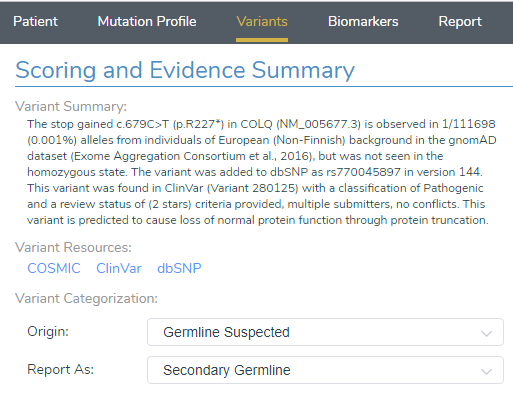
Using VarSeq, variants were first filtered based on quality control metrics such as read depth and genotype qualities to rule out false positives. The following variants were then filtered for their presence in ten common CMS genes, which was applied using the Match Gene List function from the computed algorithms list (Figure 1). The ACMG classifier was then used to isolate a single stop gained c.679C>T variant in the COLQ (NM_005677.3) gene, which was pulled into VSClinical to be evaluated according to the ACMG guidelines (Figure 2).
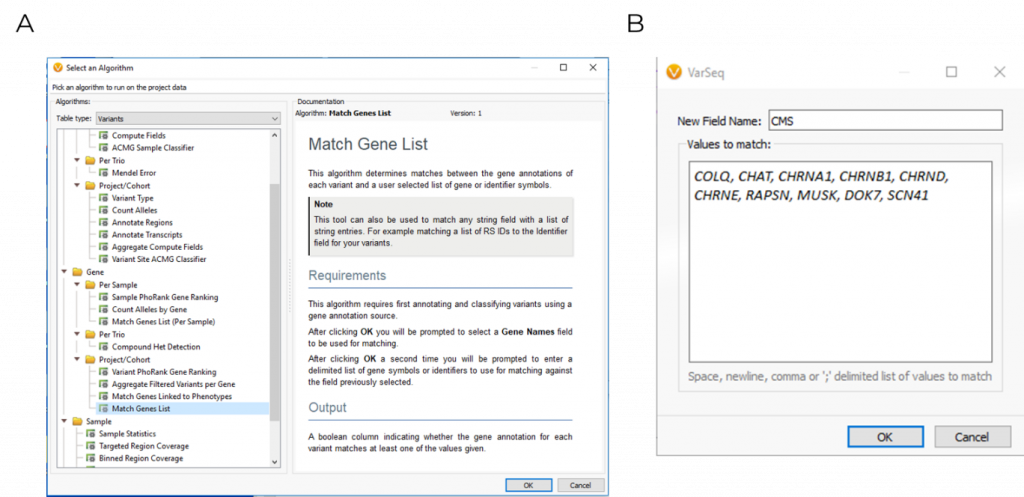
VSClinical ACMG
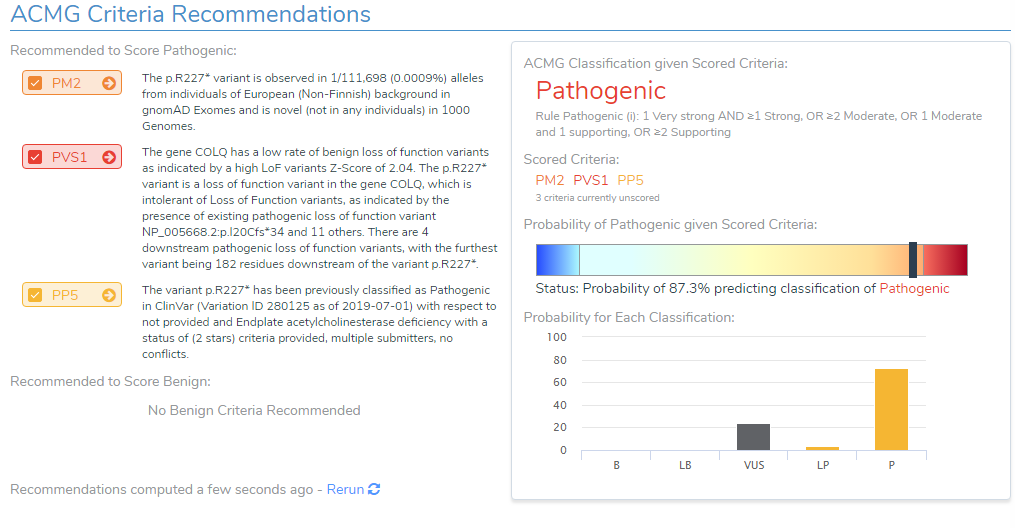
After loading the COLQ stop gained variant into VSClinical, the ACMG auto-classifier recommended to score this variant as Pathogenic for the following reasons:
- The variant is observed at an extremely low frequency in European (Non-Finish) populations (<0.0009%) according to gnomAD exomes and 1000 genomes. Due to the absence of this variant in population catalogs, the moderate criteria PM2 was applied to the evaluation (Figure 3).
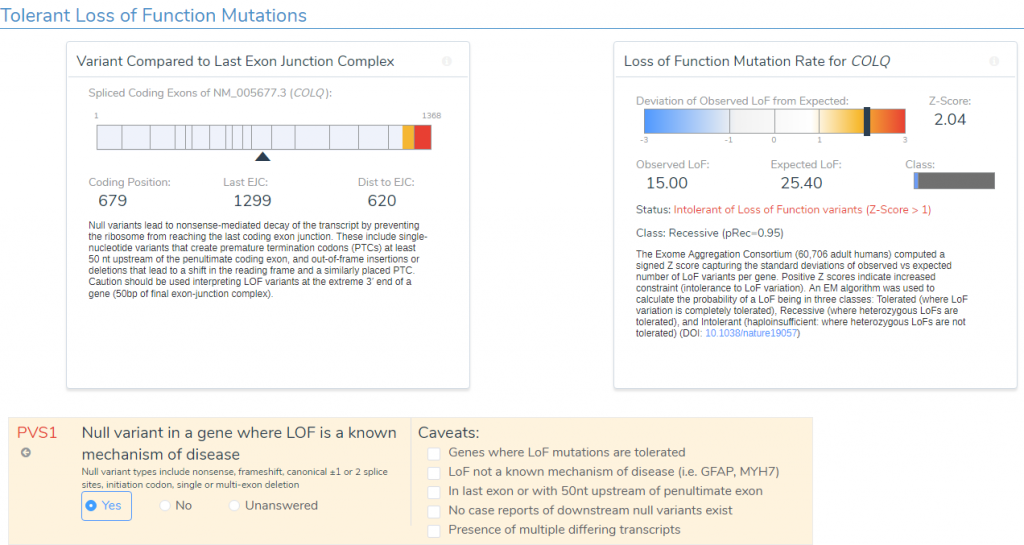
- The variant occurs 1299bp from the last exon junction complex, leading to nonsense mediated decay of the transcript and the COLQ gene is intolerant of LOF variants as indicated by the high Z-score and other downstream pathogenic variants (Figure 4). Using this evidence, PVS1 was applied to the interpretation.
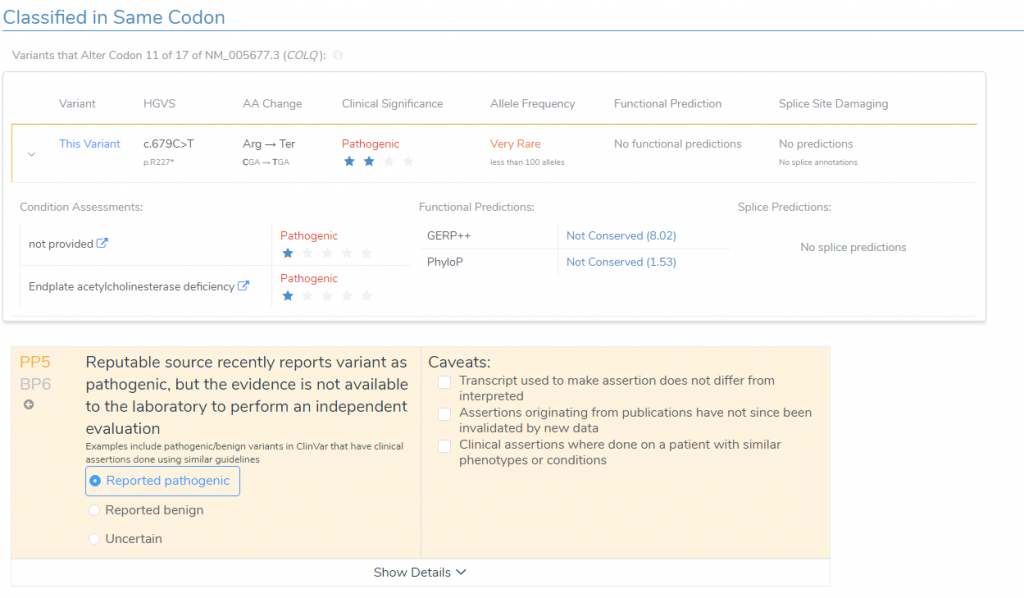
- According to ClinVar Assessments, GeneDx classified the same variant as Pathogenic that received a 1-star review status for endplate acetylcholinesterase deficiency (Figure 5). This applied PP5 to the variant evaluation.
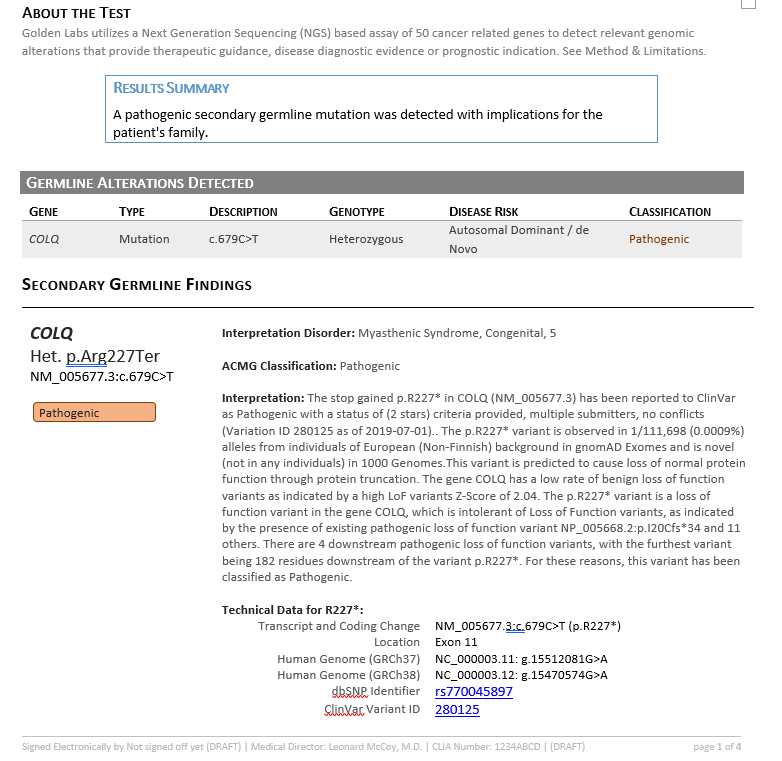
Together, VSClinical helped consolidate all available variant evidence and provided the recommended scoring criteria to reach a final variant classification of Pathogenic according to the ACMG guidelines. In addition to the interpretation, the Search Google feature provided literature indicating therapeutic recommendations including albuterol and ephedrine (3,4). This interpretation and relevant literature was then saved to the assessment catalog, or the internal database of previously classified variants, and a report was created using all information pulled from the ACMG evaluation (Figure 6).
ACMG Report
Summary and Conclusion:
An infant was diagnosed with congenital myasthenic syndromes (CMS) and genetic testing revealed a stop gained c.679C>T variant in COLQ gene (NM_005677.3). In VSClinical, the variant occurred at a low frequency in population catalogs, lead to nonsense mediated decay of the transcript and was previously classified as pathogenic from GeneDx. Furthermore, using the intuitive searching features, we identified publications that indicated variants in the COLQ gene can be treated with targeted or combination therapies of ephedrine and albuterol, which have been shown to be beneficial in several individuals (3,4). Ultimately, VarSeq is an all in one platform that allows users to start with the VCF file, filter down to clinically relevant variants, evaluate using the ACMG guidelines, and render a clinical report in a highly automated fashion.

Works Cited:
- Angela Abicht, et al. Congenital Myasthenic Syndromes. Gene Reviews.
- Violeta Mihaylova, et al. Clinical and molecular genetic findings in COLQ-mutant congenital myasthenic syndromes. BRAIN, Volume 131m Issue 2, March 2008, 747-759.
- Liewluck T, et al. Beneficial effects of albuterol in congenital endplate acetylcholinesterase deficiency and Dok-7 myathenia. Muscle Nerve, 2011 Nov; 44(5):789-94.
- Besteu-Cardiel M, et al. Congenital endplate acetylcholinesterase deficiency responsive to ephedrine. Neurology. 2005 Jul 12;65(1):144-6.
Check out some of the other blogs in this series:
- Variant Interpretation with VSClinical: Clinical Example for Congenital Indifference to Pain
- Variant Interpretation with VSClinical: Evaluation of an X-linked recessive mutation
- Variant Interpretation with VSClinical: Evaluation of Hypertrophic Cardiomyopathy
- Variant Interpretation with VSClinical: Huntington’s Disease (HD)
- Variant Interpretation with VSClinical: Non-Small Cell Lung Cancer
- Variant Interpretation with VSClinical: RET-KIF5b Gene Fusion