When doing next-generation sequencing (NGS) analysis in VarSeq, the fundamental goal is to develop efficient ways to filter through your NGS data. If you are just getting started with Varseq, a pre-designed project template can really come in handy for variant filtering! This blog series will cover a number of template design recommendations for variant filtering on data types ranging from single samples to large cohorts of whole exomes or whole genomes. To get started, let’s review one of our templates suitable for variant filtering on a single clinical or research sample.
In any workflow, whether you have gene panel, whole exome (WES) or whole-genome (WGS) data, or whether your variants are germline or somatic, there are some standard filtering strategies that can be deployed. The image below (Figure 1) is a high-level view of a standard variant filtering strategy and project layout for a single exome sample with 28,682 germline variants imported from the VCF.
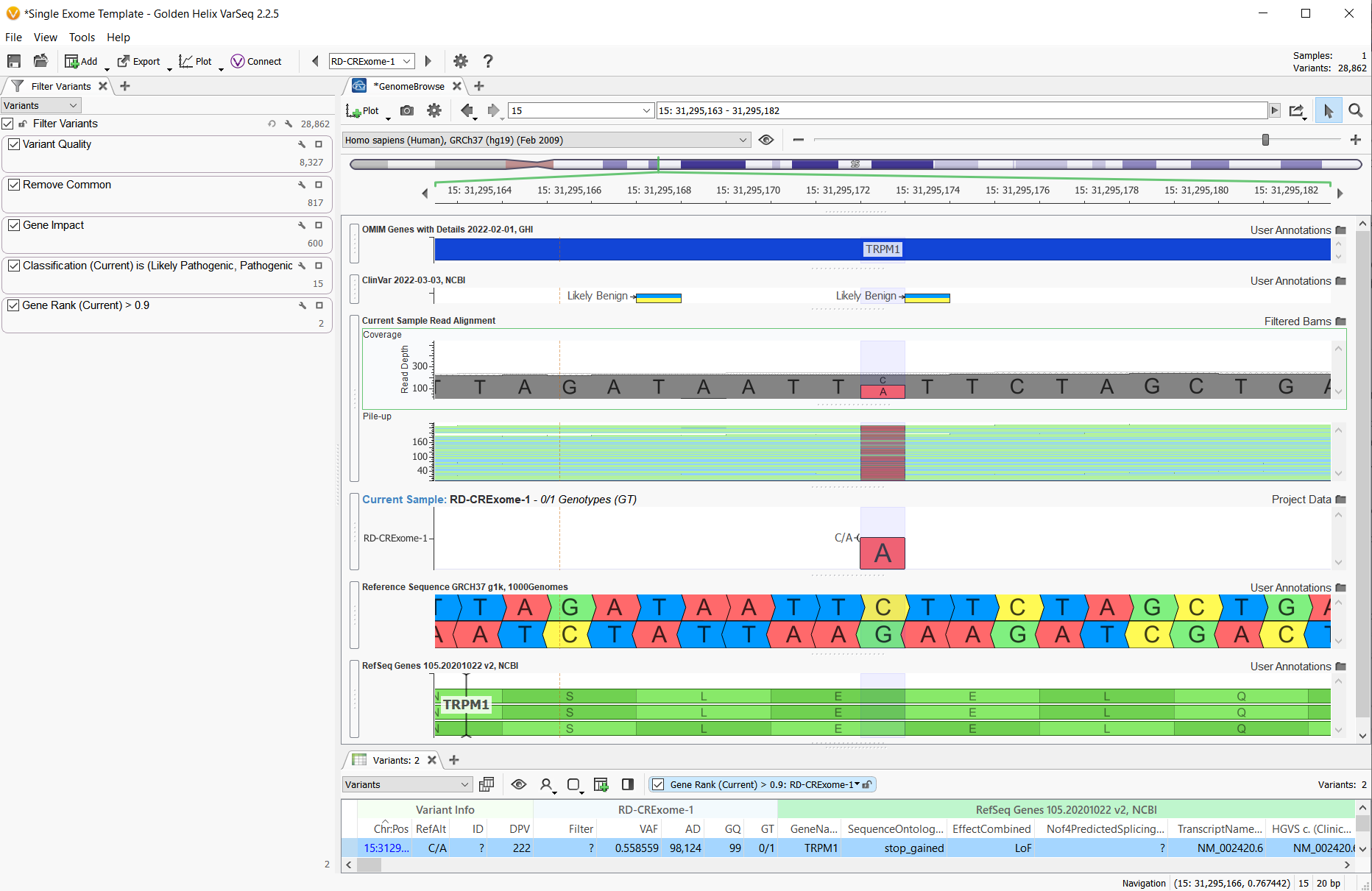
In a classic single sample variant filtering strategy for germline variants, we begin by filtering on fields from the VCF. The “Variant Quality” set of filters is used to identify variants with sufficient Read Depth and Genotype Quality, for example, and in so doing filter out potential false positives, taking the number down to 8,327 (Figure 2).
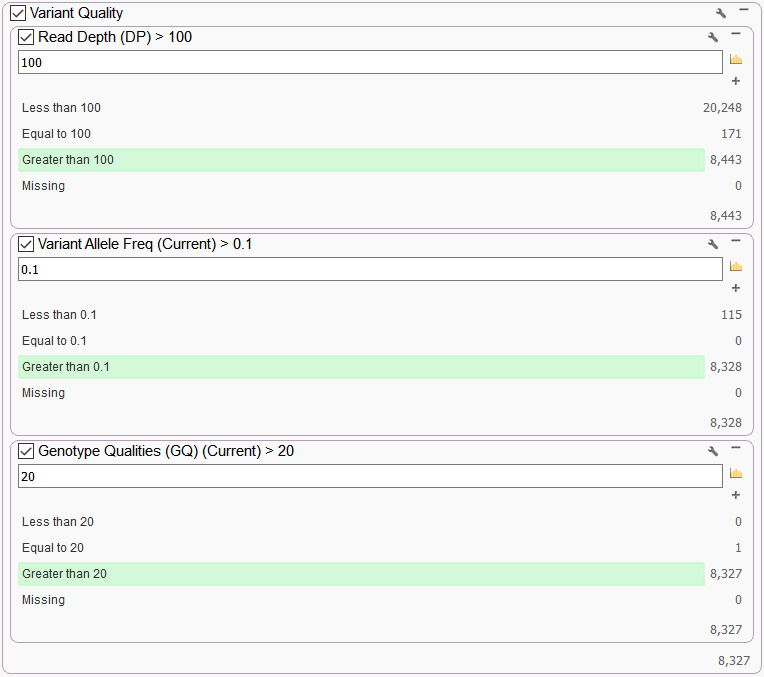
Next, we use the “Remove Common” set of filters to identify the rare variants in our sample. Here we filter on population frequency databases such as gnomAD Exomes and NHLBI to remove those variants that are considered to be common in the general population and less likely to contribute to disease. We use the alternate allele frequency field set to 0.01 and 0.99 thresholds to capture rare alternate alleles and those cases where the corresponding reference allele is actually rare. We also use “missing” to capture any rare variants that don’t exist the population catalogs. This substantially narrows the number of potentially clinically relevant variants to 817 (Figure 3).
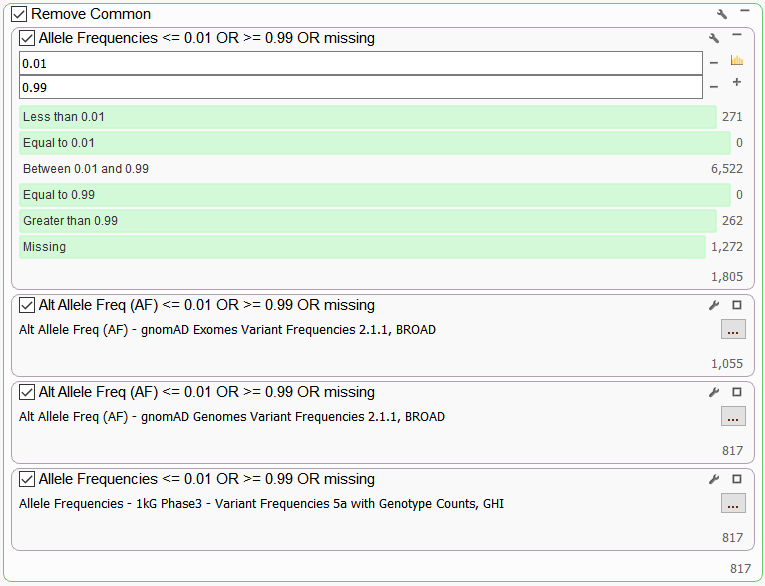
We also want to isolate any variants that affect the gene product in specific ways. While we’re not focusing on any one particular gene, we use the “Gene Impact” cards to filter out variants that are going to impact the gene in some way that we care about, for example, loss of function (LOF) or missense mutations OR those that are near enough to a splice site to cause concern. These filters can be sourced from the RefSeq track Effect (Combined) and Distance to Splice Site fields. For this particular variant filtering strategy, we leverage the OR logic option that is available with any filter container on clicking the wrench icon at the top of the container to create parallel filters.
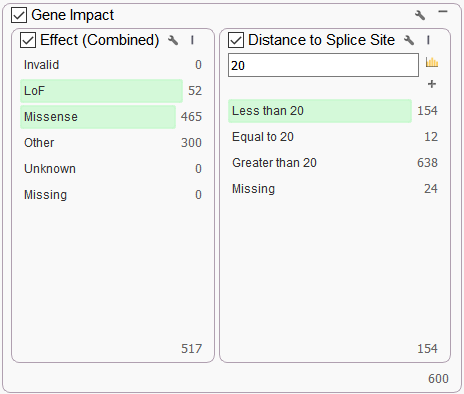
One extremely powerful filter that Golden Helix provides is the ACMG auto classifier which can be used to isolate any variant that would be pathogenic or likely pathogenic according to the ACMG guidelines (Figure 5). This is an algorithm that synthesizes data from multiple databases and is unique to our software.

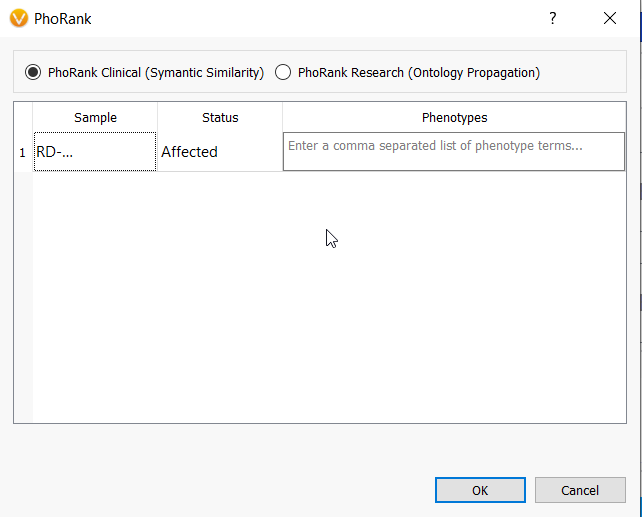
Another hugely beneficial tool for focusing your analysis is to leverage VarSeq’s PhoRank algorithm. This algorithm assigns scores from 0 to 1 to each gene based on the relationship between the gene and some user-defined phenotype(s). Here, the higher the score, the stronger the relationship to the disorder so we typically set the threshold to 0.9, but this can be edited according to user preference.

When deployed in a template, the user will be given the opportunity to input the phenotype or disorder that is relevant to their particular sample once the variants are imported.
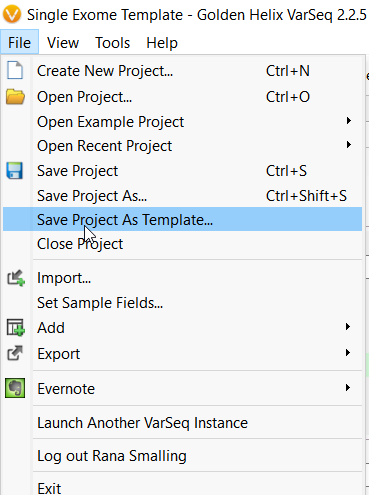
Once edited, a user can also save their changes as a new template for use with future samples.
Using this variant filtering strategy, we were able to identify 2 potentially clinically relevant variants that were of good quality, rare, impacted genes that are strongly related to our phenotype of interest and classified as either pathogenic or likely pathogenic according to the ACMG guidelines. This template is designed for use with a single sample and you can access this and other templates by reaching out to us at support@goldenhelix.com. These templates are a great starting point for your NGS analyses, and of course, can be edited to better suit your needs.
If you found this blog post useful, you may be interested in exploring additional blogs and webcasts available on our website. Thank you for reading, and if you have any questions or comments about the contents of this article please reach out to us at support@goldenhelix.com.
Great Article! Really Very insightful and fresh.
Thank you! We’re very glad you found this article insightful!