VarSeq 2.2.0 was released today and this a stable release full of upgrades and polishes. Some of the newer features include the ability to store and include AMP Cancer assessment catalogs on VSWarehouse, quicker accessibility to common annotations plotted in GenomeBrowse, and the addition of all of our standard templates for the GRCh38 genome assembly.
Many of the polishes were geared at speeding up workflows and doing a better job of handling multi-allelic variants. The workflow was also improved in VSClinical by providing more notifications when default transcripts are changed when classifications in different catalogs has changed, and even when the maximum population frequency for a variant has changed.
There were also updates to the Blinded Interpretation option with the ACMG Guidelines so that a multiple user organization can better compare evaluations from different users without having previous interpretations influence analyses.
New Features
When running the Annotate Transcripts algorithm, a new field “Coding Diff” displays the amino acid coding change. For example, cAg/cGg for a Gln/Arg missense.
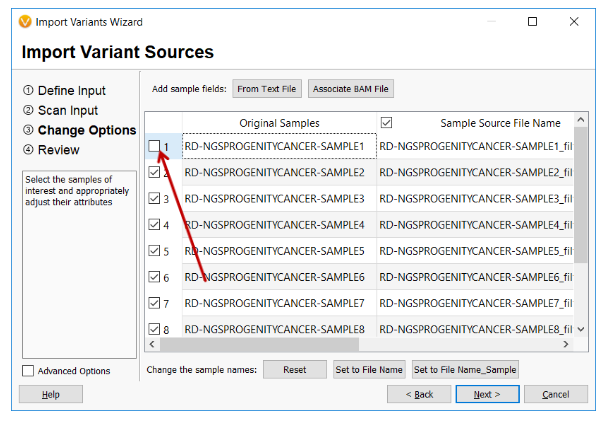
Samples and their associated variants will now be removed from a project on import if the sample is unchecked in the Import Wizard and the variants do not exist in any other sample set. This works regardless of whether the import files are per-sample or multi-sample variant files.
A revert/undo icon was added to the top of the filter chains to allow the previous filter move to be reverted.
The Count Alleles algorithm now has the option to list the names of a limited number of samples that contain heterozygous or homozygous genotypes.
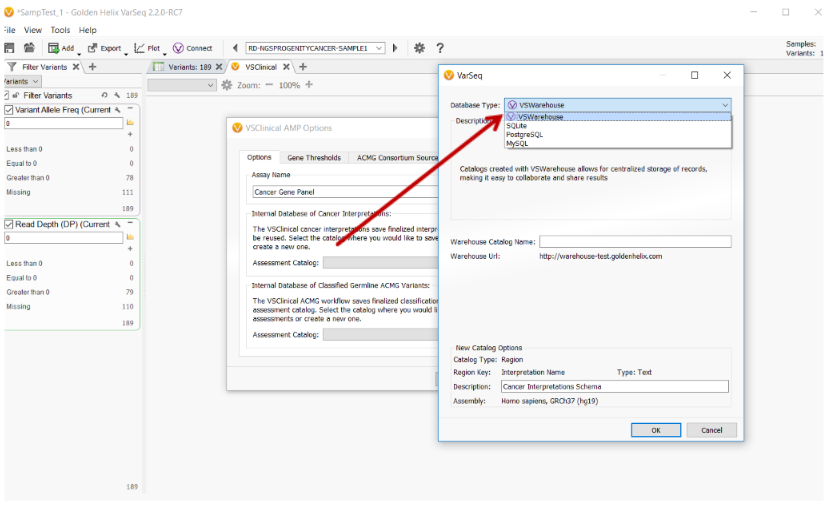
The assessment catalogs used to store the Cancer Interpretations for the VSClinical AMP Guidelines can now be created and stored on VSWarehouse.
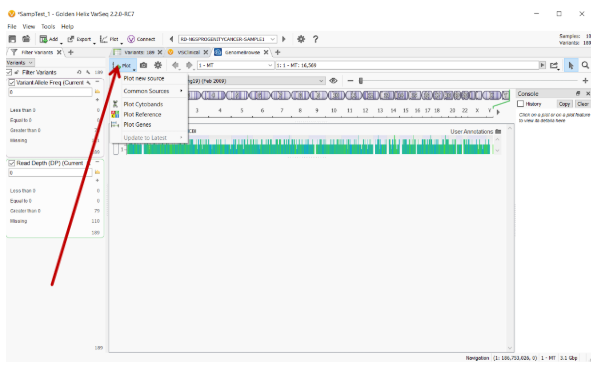
The Plot button in GenomeBrowse has been upgraded to a dropdown menu that allows users to quickly access commonly plotted annotation tracks and update plotted tracks if more current track versions are available.
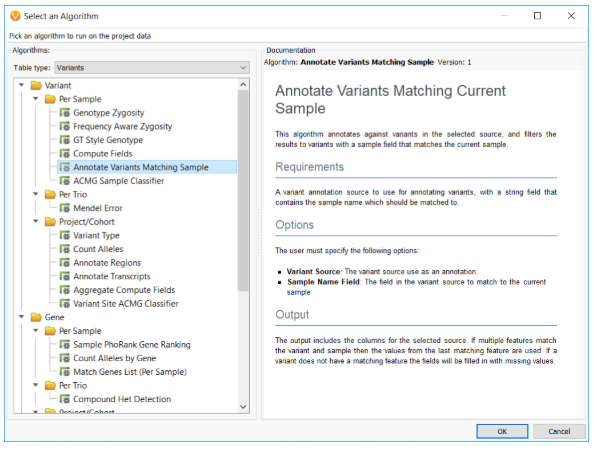
A new algorithm called Annotate Variants Matching Sample was introduced that allows sample-specific variant annotating and is especially useful when annotating with sample-specific assessment catalogs.
VarSeq has added standard project templates for the GRCh38 genome assembly.
Bugs Fixed
- The latest version of gnomAD 2.1.1 splits out multi-allelic variants into their own individual records, not preserving the multi-allelic form. This meant sites with two common non-reference alleles were not being annotated by gnomAD 2.1.1 and filtered out. A new multi-allele aware variant annotation mode was added to support this representation and multi-allelic variants will now match records for the individual alleles in gnomAD.
- Editing the sample table and modifying the parent fields did not update all dependent algorithms, but now modifying the parent relationships between samples will update algorithms that rely on these relationships like Mendel Error.
- Adjusting the color of an already established variant set did not adjust the color of the variant set column immediately but does now.
- Switching the filter chain between table types (like from Variants to Coverage Regions) was disabling the previous filter type.
- The project link created when using the Project Name as an assessment catalog field was broken.
- Exporting multiple sample files to XLSX file format caused a crash. This has been fixed to allow multiple samples to be exported to XLSX format at the same time.
- The import wizard layout has been updated on multiple pages and especially the finalize page to better handle the many options on different size screens and platforms. The many import options are displayed in expandable and collapsible menus to conserve space. Also, a few help links to the manual on advanced options were fixed.
- When annotating coverage regions and CNVs with genes, the percent CDS coverage field was incorrectly computing the percent of the entire gene instead of the coding regions of the gene.
- In the assessment catalog import data wizard, the layout of the fields has been fixed so that it cannot be minimized beyond comprehension.
- Certain tracks being plotted in GenomeBrowse with missing file paths were taking time to error out, but this has been updated to error out immediately and informs the user of the missing file path.
- In GenomeBrowse, when coverage was being computed for a BAM file and the user pauses the computation, one file is created on resuming and not multiple versions.
- Using the Data Annotation Convert Wizard with the left-align feature, we require a local copy of the reference sequence but did not handle the case when it was not present. Now the Convert Wizard shows a message to the user to download the required reference sequence if needed.
- The following changes have been made for VSClinical:
- When adding multi-allelic variants from the table, only the alleles present in the current sample are added to the evaluation. In general, a lot of work went into improving the complex cases of handling multi-allelic variants in this release.
- When switching which transcript a variant is based on, the Variant Summary and Auto Classification will now update to reflect the changed HGVS.
- Previously, VSClinical always requested CADD annotations remotely, even when there was a local copy of CADD. This has been fixed to recognize the downloaded track.
- Similarly, locally downloaded Multiple Sequence Alignment sources were not being used in favor of remote access. They will now be used.
- Malformed ACMG records were causing errors. These may have been set by manually adding variants to an ACMG catalog. This has been fixed to more robustly handle malformed records.
- The option to manually add variants in the ACMG Guidelines workflow now prevents the user from selecting a variant that is not in a gene for analysis.
- VSClinical now does a much better job of handling projects with annotation sources that are incomplete or have missing input.
- The ACMG Classification algorithm would not consider variants saved in the configured catalog as “Uncertain Significance”. It now overwrites the auto-recommendation with the last saved user classification, including a VUS classification.
- The Variant Evidence card in the ACMG Guidelines allows users to switch between GRCh37 and GRCh38 assemblies and if the liftover can’t be performed, an error message is now shown.
- Certain missense variants that lacked a missense badness score were showing errors in the ACMG Workflow. This has been corrected to allow these variants to be evaluated.
- The following changes have been made for VSClinical AMP Workflow:
- In some cases, the thresholds for the Coverage Regions plots were reset to the default values on the project open. This has been changed to keep the preferred values.
- In some cases, intronic variants were not reported as intronic in the “Location” field in the reports tab.
- If a specific tumor type had an interpretation saved for a Gene Summary, it was not being preferred over an interpretation for all cancers for new Biomarkers.
- The Wild-Type variant dialog was not showing the most up-to-date variant count for the selected gene, and now updates correctly.
- In the Null Variants in Oncogenic Gene section of the Variants tab, the card displaying supporting evidence for LoF variants in the gene did not update to match the current gene.
Polishes
- Performance of using VarSeq assessment catalogs with VSClinical or directly has improved for the use case of multiple users using an SQLite file-based catalog on a shared networking drive, especially during concurrent access.
- When an algorithm or annotation source is updated, new fields will be hidden in existing tables by default. You can right-click on the table source header or open the Visibility dialog to adjust visible fields.
- Empty columns that were created when using the Compute Fields algorithm on a subsequent source are now removed from the computation preview.
- The overlapping transcripts algorithm and the annotate transcript algorithm both utilize the same gene preferences file for continuity.
- When naming new variant sets, numbers can now be used in the second position of the naming Initials.
- When opening a custom project template where an input annotation source is not found, there is now an option to delete the algorithm that uses the missing annotation source on import.
- VarSeq more efficiently authenticates with VSWarehouse when performing assessment catalog operations.
- The Convert Wizard has a new option when lifting over variant tracks to adjust counts on known fields when the reference and alternate alleles are flipped in the new assembly. This feature is checked by default to match the previous behavior.
- On Linux, opening external programs such as the web browser or folder explorer now works on a large variety of Linux flavors and systems.
- Now when associating BAM files to samples, the file name closest to the sample name will be selected first.
- Tracking the current connected warehouse is now more robust to differences in inclusion of trailing slash in URL.
- The ability for users to control memory settings has been adjusted to provide Low, Medium, High, and Custom options.
- In GenomeBrowse, coverage statistics do not automatically start computing until the user requests it. Also, a recent regression in the performance of computing coverage in the previous VarSeq version has been fixed.
- The table icon on GenomeBrowse plots now acts as a toggle to show and hide un-docked table views of the current plot data.
- Switching the assembly in the assembly dropdown menu in GenomeBrowse used to switch the assigned assembly for the project. This caused confusion and has changed so that assembly switching in GenomeBrowse is independent of the project assigned assembly.
- The following polishes have been made for VSClinical:
- You can now start an evaluation even without running the ACMG Sample Classifier algorithm. In general, less dependencies are made on the variant table to ensure the latest algorithms and state of previous assessments are always represented.
- Warnings and notifications have been added to ACMG Guideline classifications to show when previous classifications were last saved, if any scoring criteria has changed since the previous classification, if the ClinVar classification record has changed, and if the maximum population frequency has changed since the previous classification was made.
- The ACMG Guidelines will also show a Special Considerations notification when a variant overlaps multiple genes or transcripts or scoring seems to have been left in an incomplete state regarding clinical or functional evidence.
- The Blinded Interpretation option in the ACMG Guidelines has been updated to fully blind previous classifications and interpretations to the current user. The resulting evaluation starts off as if the variant was being seen for the first time.
- The Gene Region and Mutation Profile table has been updated to include the HGVS amino acid description and have a “Sources” column that makes it clear which entries are coming from ClinVar, your internal catalog or both. Similarly, tooltips on each column break out the details coming from each source. In the AMP workflow, CIViC records are integrated into this table as another unique source. Optionally configured Consortium Sources will similarly be integrated.
- The Related or Nearby Variants section of the Studies tab in the ACMG Guidelines has been enhanced so variants previously seen can be clicked to show that variants Historical Interpretations dialog.
- The Download Dependencies screen for the AMP and ACMG Guidelines does not appear every time a new tab is opened if initially skipped or all required dependencies are downloaded.
- The ACMG criteria that were scored during a previous classification in the ACMG Guidelines will now be set as scored when a new evaluation for the previously scored variant is opened.
- The ACMG Options dialog now displays the assessment catalog database type and location with a destination link where just the file path to the assessment catalog was displayed before.
- The Variant Finalization screen in the ACMG Guidelines now has an ‘Update Previously Classified’ option to request the latest saved classifications information from the assessment catalog for the current variants in the evaluation.
- Now when switching between variants in the ACMG Guidelines, the view of each expanded section is preserved to more easily compare sections between variant analysis.
- Variant cards that allow users to switch between GRCh37 and GRCh38 views will now automatically open on the default assembly of the project.
- The Historical Interpretations dialog now displays more details about the previously scored variants in the list.
- The Historical Interpretations section of previous variant classifications in the ACMG Guidelines now shows the most recent classification first and is sorted in reverse chronological order.
- The Variant Summary for variants that extend beyond gene boundaries has been improved to better emphasize the upstream and downstream impact.
- For variants that overlap 2 or more genes, only 1 gene will be shown in the Variants to Evaluate list at the start screen of the ACMG Guidelines. Once the evaluation is created, the variant can change which gene is being used in the evaluation from the change transcript dialog. The fact that the variant overlaps two genes is placed in the new Special Considerations section.
- Phrasing has been adjusted in the ACMG Guidelines to reserve the term, “Finalize” for samples and evaluations. Before, individual variants could be finalized, but now they are only, “Saved”.
- Saving a variant in the current should not change the number of “Previous Samples” reported seen for that variant in various contexts. We ensure that the current sample is never counted in this count.
- The following polishes have been made for VSClinical AMP Guidelines:
- The version of COSMIC used in the Somatic Catalogs section of the Variant Annotation tab is now displayed.
- When adding a variant that is missing a protein notation, the coding notation is used.
- A warning message now exists to remind users that the tumor type needs to be set before adding biomarkers to the AMP Guidelines.
- Now when adding variants to the mutation profile, the Oncogenicity/Pathogenicity classification computed informs how by default the variant will be reported. Benign/Likely Benign variants will be set to “Don’t Report”.