With the latest release of VarSeq, we have made significant updates to our handling of the interaction of variants and genes. This includes the support for non-coding transcripts, improved splice site predictions, and updates to gene and transcript annotations.
We received several questions regarding how decisions are made in the software regarding genes and transcripts with these gene-related changes. This is a great opportunity to review the powerful Gene Preferences system in VarSeq, which ultimately lets users and labs control exactly how specific genes should be handled when it comes to variant annotation and scoring using the ACMG and AMP workflows of VSClinical.
Saving Preferences for Genes
While the genomic annotation library curated by Golden Helix is second to none, gene annotations alone are insufficient to prepare a variant for a clinical report. When it gets down to the details of interpreting a variant following a scoring and classification system, specific opinionated choices about how to report a gene must be made, and those preferences remembered. The VarSeq algorithms and VSClinical workflow engine look up these properties from two locations, the User Gene Preferences and the System Gene Preferences. The user’s preferences will always take precedent and are shared amongst multiple lab users using the same folder location as assessment catalogs (often configured as a network share). The system gene preferences file is bundled as part of the VarSeq program and updated with the software.
The following are the preferences that can customized at the gene level:
- Transcript: The preferred transcript to use, overriding the default choice chosen by a heuristic
- Inheritance Mode: The disease inheritance model to be used: Dominant vs. Recessive
- Disorder: The disorder to report for this gene (along with linked OMIM / MONDO identifiers)
- WildType for Tumors: A Cancer workflow specific option to specify which tumor types indicate this gene as clinically significant in a Wild Type state.
How Gene Preferences are Used
There are several contexts in the VarSeq annotation and filtering workflow and the VSClinical interpretation workflow that leverage the gene preferences system. Because you can customize gene preferences, the clinical lab defines the behavior in these contexts when defining and validating their clinical test.
Transcript Annotation: The VarSeq gene annotation algorithm deals with a lot of complexity when describing the interaction between project variants and overlapping transcripts. While there is a detailed table called “Transcript Interactions” that provides the combination of every alternate allele for a variant combined with every transcript. Ultimately, clinical reports report on a single transcript. This “Clinically Relevant” transcript is something VarSeq will select and display in the summary columns set of the gene annotation algorithm. Along with the transcript, we also provide the HGVS coding and protein notation, the sequence ontology, and other relevant gene annotation details, including splice site predictions. If a gene has no entry in either the system or user gene preferences file for a preferred transcript, a heuristic is used to select a clinically relevant transcript (more details in a subsequent blog post).
ACMG Classifier: The scoring of the ACMG guideline criteria codes can be automated for the criteria based entirely on annotations and bioinformatically derived attributes of variants. The ACMG Classifier will evaluate a variant based on the clinically relevant transcript selected by the gene annotation algorithm in the project. Also, the Inheritance of the gene is reported and used when evaluating certain criteria. The evaluation of whether a variant is rare or common uses different thresholds (stricter) for dominant model gene inheritance versus recessive (or unknown).
VSClinical Recommendations: After the annotation and filtering VarSeq workflow, certain high-quality variants of interest will be taken forward to interactive scoring and interpretation and VSClincial. Both the ACMG and AMP (Cancer) workflows do up-to-date variant scoring when the variants are added to an evaluation. At this time, the preferred transcript, inheritance model, and any previously saved disorder (and identifiers) are looked up for the variant and used to perform the variant scoring and fill into the interpretation. The cancer workflow will also lookup whether the current patient’s tumor type matches any marked as relevant wild types in the gene preferences.
Updating Your Gene Preferences
There are a couple of places in the VSClinical workflow where a gene preference can be updated. When a variant interpretation is saved, either as an ACMG germline variant or an AMP somatic variant, if the transcript or currently selected disorder differs from the current gene preference, the bottom of the save dialog will display an option to update the saved gene preference of each of these properties when the variant interpretation is saved.

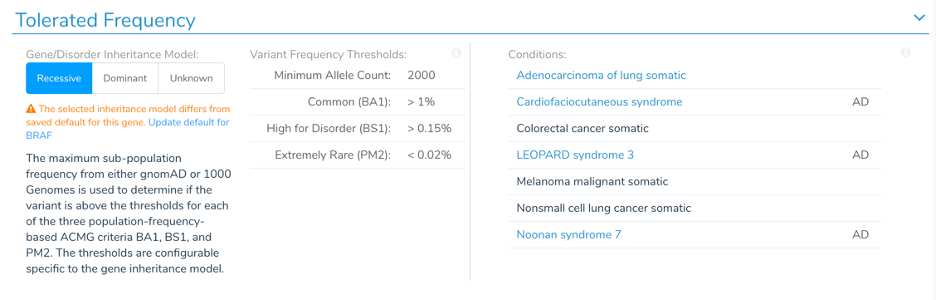
The mode of inheritance preference is updated in the Tolerated Frequency section of the ACMG guidelines. You can change and save the Inheritance value directly and see the implications on the frequency thresholds used for BA1, BS2, and PM2. On the right, the associated conditions from OMIM and their annotated inheritance model are provided for reference.
Powerful and Transparent
The Gene Preference system in VarSeq allows labs to control the critical choices of which transcripts are selected for annotation and reporting. GenePreferences.gene-pref can be found by going to Tools > Open Folder > Assessments Catalog Folder. It is a structured text file and can be browsed to review the currently saved gene preferences. In the future, we plan to add more direct management of these preferences.
In the latest VarSeq release (2.2.3), we updated Gene Preferences’ systems to reflect the latest changes to the RefSeq transcript and supporting gene annotations, OMIM disorder catalog, and ClinVar lab submissions. To learn more about how these data sources are used to create the default set of Gene Preferences that come shipped with VarSeq, check out the next blog post in this series!