Sentieon develops bioinformatics secondary analysis tools to process genomic data with high computing efficiency, fast turnaround time, exceptional accuracy and 100% consistency. These features are what led to the partnership of Golden Helix and Sentieon to provide users with a comprehensive solution for genomic data analysis. This blog post gives readers a more detailed understanding of the top five features of Sentieon. If these features are of importance to you, we suggest testing it out via a complimentary trial!
Process genomics data with high computing efficiency
The Sentieon tools are drop-in replacements/improvements for BWA/GATK/MuTect/Mutect2. They are easily scalable, easily deployable, easily upgradable, software-only solutions. The Sentieon tools achieve their efficiency and consistency through optimized computing algorithm design and enterprise-strength software implementation and achieve high accuracy using the industry’s most validated mathematics models.
Fast turnaround time
The Sentieon Genomics tools provide an optimized reimplementation of the most accurate pipelines for calling variants from next-generation sequence data, resulting in more than a 10-fold increase in processing speed while providing identical results to best practices pipelines. There is an excellent paper published in bioRxiv that explains this in detail.
Exceptional accuracy
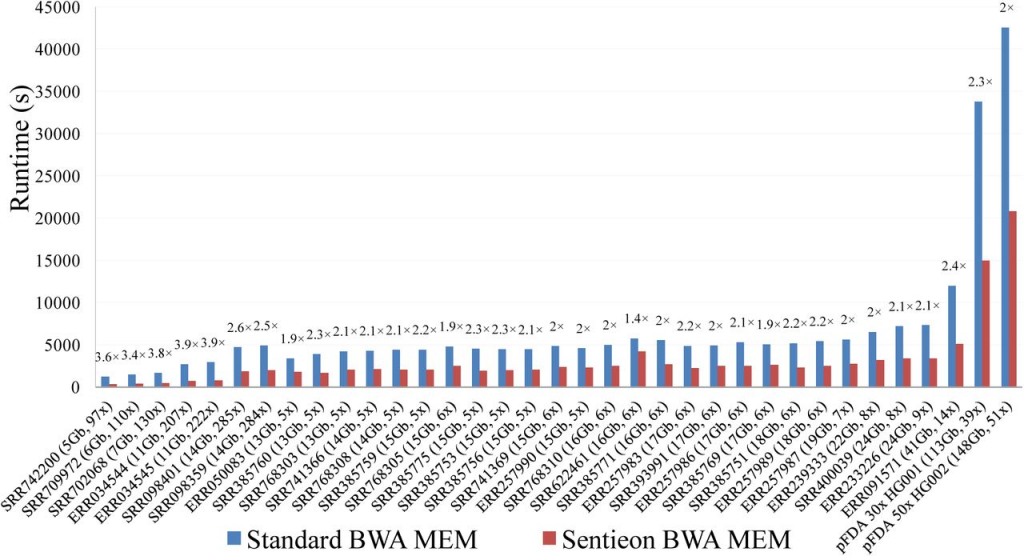
Sentieon provides an optimized implementation of BWA resulting in an average 1.9x speedup while producing identical alignments. Sentieon DNAseq and the GATK Best Practices Pipeline were run on seven whole-exome, 78 low-coverage, and two whole-genome samples. Among all samples, the Sentieon DNAseq pipeline resulted in an average 36x improvement in runtime relative to the GATK Best Practices pipeline (figure 1). The performance improvements were most notable for the indel realignment, base-quality score recalibration, and HaplotypeCaller variant calling stages where Sentieon’s tools improved runtimes by an average of 56x, 46x, and 30x, respectively (figure 2).

100% consistency
No matter how many times you process and analyze the same the data, the produced end data will always have the same results.
Customizable with scripting
If you need to run multiple lanes or a single read you can customize a script to run to produce the most accurate results. If you are producing 1 – 100+ FASTQ reads, you can batch this process by giving Sentieon a source directory where the sequencers’ output files are produced and a destination where your BAM and VCF files will reside for tertiary analysis. Grouping each sample in their own project folders.
I hope this brief overview explains the significant speed advantages Sentieon provides over other existing methods. As I mentioned in the beginning, if these features are of value to you, we encourage you to get a demo or trial of Sentieon to see how it works for you. You can request either of these by emailing our team at info@goldenhelix.com!