SVS 8.9.0 was released on August 19th and features a new GBLUP by Bin feature and a new utility to find the LD scores of markers and categorize them into bins, along with several mixed-model upgrades and many other upgrades, fixes, and polishes.
The two new features LD Score Computation and Binning and Compute GBLUP Using Bins, while they can be used separately, may be used together to study polygenic trait heritability with a better resolution than using our already-existing GBLUP feature. Wainschtein (Wainschtein, P., et. al., (2019) ‘Recovery of trait heritability from whole genome sequence data’. BioRxiv preprint https://www.biorxiv.org/content/10.1101/588020v1.full) has used this technique to recover heritability for height and body mass index (BMI) from whole-genome sequence (WGS) data on 21,620 unrelated individuals of European ancestry equal to levels (0.79 for height and 0.40 for BMI) suggested by pedigree data.
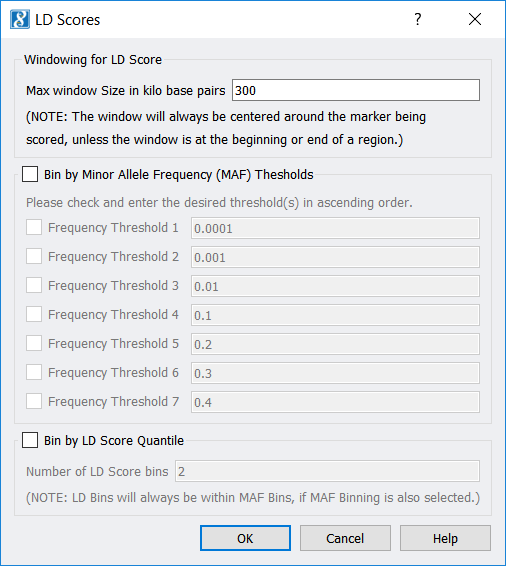
LD Score Computation and Binning
This feature outputs LD scores and MAF for your marker data and can be used to categorize (“bin”) your markers according to LD score, MAF, or a combination of both.
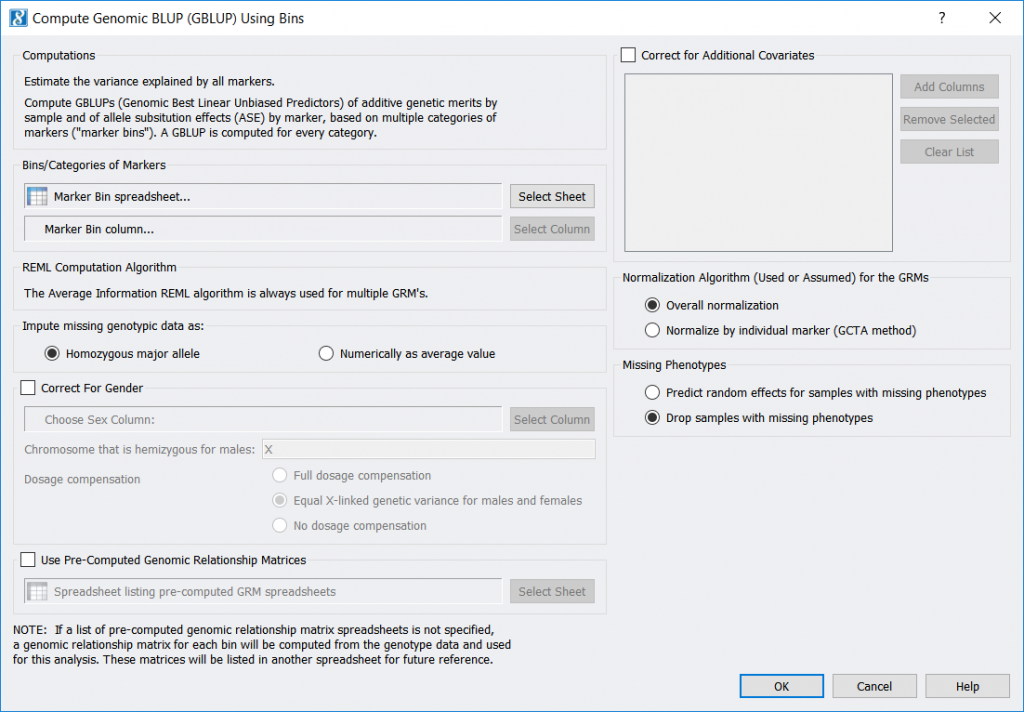
Compute GBLUP Using Bins
Compute GBLUP Using Bins estimates your markers’ polygenic effects based on using separate genomic relationship matrices for each of several categories of markers займ онлайн с 18 лет. One of its inputs is a marker-categorizing (“binning”) spreadsheet for your markers, as can be generated by the LD Score Computation and Binning feature (or created any other way, such as using a marker’s chromosome as its category).
Additional Improvements
Other mixed-model improvements include:
- K-Fold Cross Validation has a new option which allows creating sample subsets in a “sampling with replacement” mode.
- Predict Phenotypes from Existing Results may now be used on output from GBLUP or from GBLUP using bins.
- “Predicting random effects for samples with missing phenotypes” can now be selected for more use cases.
- If the Average Information REML algorithm (used by GBLUP and GBLUP using bins) is found to be not converging, the algorithm is stopped, and the most informative possible output is presented.
Other improvements include:
- The liftover functionality of VCF Import is now available.
- The family data feature of VCF Import now imports the data as a pedigree spreadsheet.
- Many other minor polishes and documentation improvements have been made.
New Features
- A new feature, Genotype > Quality Assurance and Utilities > LD Score Computation and Binning, has been added that
- Determines an LD Score for every marker,
- Finds the minor allele frequency (MAF) for every marker, and
- (Optionally) categorizes (or “bins”) every marker according to its LD Score, its MAF, or a combination of LD Score and MAF.The base pair window size for computing LD Scores may be selected, as well as MAF thresholds for binning (if MAF binning has been selected).
- A new feature, Genotype > Compute GBLUP Using Bins, has been added which performs Genomic Best Linear Unbiased Predictors (GBLUP) using markers that have been binned into categories, with a separate Genomic Relationship Matrix (GRM) being created and used for every category.The outputs of this feature correspond with the outputs of Genotype > Compute Genomic BLUP (GBLUP):
- If computed dynamically, a GBLUP Genomic Relationship Matrix (GRM) for every bin (category), and a Genomic Relationship Matrix List of the GRM spreadsheet numbers.
- Binned GBLUP estimates by sample This spreadsheet includes the total estimated random effects, the estimated random effects corresponding to each GRM, and (optionally) the predicted phenotype value for each sample.
- GBLUP fixed effect coefficients This spreadsheet shows the intercept value and the coefficients for any fixed-effect covariates you may have specified. (This corresponds to a new spreadsheet now created by Genotype > Compute Genomic BLUP (GBLUP).)
- Binned GBLUP estimates by marker This spreadsheet delineates each marker’s bin (category) name and the value of its allele substitution effect (ASE).To categorize your markers into bins, you can
- Use the new Genotype > Quality Assurance and Utilities > LD Score Computation and Binning feature (mentioned above),
- Use the chromosome name as the binning category,
- Run DNA-Seq > Variant Binning by Frequency Source from the spreadsheet menu, or
- Any other method you wish to use for categorizing markers.
- The Genotype > K-Fold Cross Validation (for Genomic Prediction) feature has a new option, Predict Only Once per Iteration. This effectively allows a mode of creating sample subsets using “sampling with replacement”.
- A new feature, Edit > Recode > Clean Up Missing Genotype Data, has been added which will convert partially-missing genotypes (such as “A_?”, ”?_B”) to the proper missing-value form, which is ”?_?”.
- A new Tools > CPU and (GenomeBrowse) Memory Settings dialog has been added to allow control over memory used for reading annotation files and for GenomeBrowse scene rendering, and also control over the number of CPU processors used. Note: This feature is distinct from the Memory Usage section of Tools > Global Product Options, which allows control of memory for dataset caching, transposing of spreadsheets, and analysis.
- Three new capabilities have now effectively been added to SVS as a result of changes (as noted in the sections below) to existing features:
- The liftover functionality of Import > Import VCFs and Variant Files is now available.
- Genotype > Predict Phenotypes from Existing Results may now be used on results from Genotype > Compute Genomic BLUP (GBLUP).
- Dominant-model or Recessive-model data (as well as Additive-model data) may now be PCA-corrected, then used as input to either Genotype > Mixed Linear Model Analysis or Genotype > Mixed Linear Model Analysis with Interactions.
Polishes
- Genome assemblies have been added and updated, including Chicken (Gallus gallus, GRCg6a), Cowpea (Vigna unguiculata, JGI_UCRv1.0), Water Buffalo (Bubalus bubalis UOA_WB_1), and Barley (Hordeum vulgare IBSC_v2).
- Several SVS tutorials have been updated to account for newer features (such as SKAT-O) and other improvements in SVS, to use newer annotation data, or to make the explanations more clear.
- The Data Source Library window now automatically refreshes after a track finishes downloading.
- Target Region CNV options and Whole Genome CNV options may now be saved as project options or global options.
- The final screen for Import > Import VCFs and Variant Files has been greatly improved to show all import parameters clearly.
- The family data feature of Import > Import VCFs and Variant Files now imports the data as a pedigree spreadsheet.
- The HapMap importer (Import > HapMap) has been updated to be able to incorporate IUPAC single-letter encodings.
- The instructional prompts, error messages, and the log messages for Edit > Recode > Recode AGCT Alleles to AB were modified to increase clarity for the user.
- The names of the menu items in the Affymetrix CNV import options have been improved to clarify what type of data is supported.
- The following improvements have been made to the Genotype > Compute Genomic BLUP (GBLUP) feature:
- The intercept value and the beta values for the covariates are now output as a separate spreadsheet, using the same format as does Genotype > K-Fold Cross Validation (for Genomic Prediction). This means that Genotype > Predict Phenotypes from Existing Results may now be used with outputs from the Genotype > Compute Genomic BLUP (GBLUP) feature.
- Predict random effects for samples with missing phenotypes may now be used with the Average Information algorithm (and thus with features requiring the Average Information algorithm).
- The user interface for Genotype > Compute Genomic BLUP (GBLUP) (and for Genotype > Quality Assurance and Utilities > GBLUP Genomic Relationship Matrix) now detects whether or not you have an X Chromosome in the marker map, and changes its defaults (and other behavior) accordingly.
- In Genotype > Genetic Correlation of Two Traits using GBLUP, Exclude the residual covariance from computations has been changed to be the default, since this invokes a version of this algorithm which more realistically represents the problem, and also is more likely to converge.
- For the Average Information REML algorithm, both for this feature and for the Genotype > Genetic Correlation of Two Traits using GBLUP feature, SVS will always strive to give useful output, even if the algorithm “did not converge”.
- The Variance/Covariance matrix from the Average Information REML algorithm is now output as an SVS spreadsheet.
- Calculation of allele substitution effects (ASE) for multiple GRM’s has been optimized.
- A few error messages (and error notes for the node change log) for the EMMA algorithm, as well as messages to note collinearity of covariates, have been clarified. In addition, many messages have been reformatted to improve their appearance.
- Integer columns are no longer allowed as gene-by-environment category variables. (This feature works best using only a limited number of categories. Integers with only a limited number of distinct values may always be converted to “strings” (categorical variables) in Edit > Edit This Spreadsheet.)
- The following improvements have been made to Genotype > Predict Phenotypes from Existing Results:
- Dosage Compensation and Individual Normalization may now be specified as inputs.
- The checking for whether the covariates listed in the fixed-effect spreadsheet are all re-specified as covariates when using this feature has been made more precise.
- Random effects will now be predicted for all samples. This is in addition to the current output of predicting phenotype values for samples with valid covariate values.
- The overall flow of dialog and execution has been improved.
- The run log for Genotype > Bayesian Genomic Prediction is now updated more often, so it is less likely that the software will appear to have stopped running when it hasn’t.
- For Numeric > CNV on NGS Target Regions, there is now a progress dialog for creating the state spreadsheet, and an optimization for creating the state spreadsheet when showing a categorical CNV state. Thus, the software will not appear to have stopped running at this step when it hasn’t.
- For Genotype > Mixed Linear Model Analysis with Interactions, outputs are now documented for the Linear Regression (fixed effects only) with Interactions sub-feature.
- The mathematics for Genotype > Mixed Linear Model Analysis with Interactions has been more fully documented.
- For the Genotype > Mixed Linear Model Analysis Multi-locus mixed model GWAS (MLMM) feature, how the p-value for a “covariate marker” is found is now documented more clearly.
- Other documentation for the Mixed Linear Model Analysis features has been polished.
- For Genotype > K-Fold Cross Validation (for Genomic Prediction), Dosage compensation is available as an option when using GBLUP.
- Genotype > Quality_Assurance > Mendelian Error Check will now handle partially-missing genotypes.
- The link to the documentation for the Illumina Genome Studio SVS plug-in, which is no longer supported, has been removed.
- The Formulas and Theories documentation sections on Linear Regression for Association Testing and on Multiple Linear Regression have been improved.
- A more specific explanation of what constitutes the “number of tests” to be used for a Bonferonni correction has been given for each of many different tests.
- For BEAGLE imputation, the Allelic R-squared and Dosage R-squared outputs are better documented.
- The hard limit for the number of samples that can be processed by Principal Component Analysis (PCA) has been removed. Additionally, the number of samples for PCA analysis that triggers a question about memory usage has been increased to 8,000.
- Internal code changes to Principal Component Analysis (PCA) have been made to slightly optimize performance and to make future changes more reliable.
- The Secure Annotation source MedGenome OncoMD is no longer supported.
Bugs Fixed
- The Genotype > Quality Assurance and Utilities > Fixation Index Fst feature was hanging up and stalling while computing confidence intervals. This has been fixed.
- The user is now prevented (with an error message) from trying to simultaneously import both VCF files that have samples and other VCF files that do not have samples. Before, SVS would try to do this, but crash.
- The liftover functionality of Import > Import VCFs and Variant Files has been fixed for SVS.
- The SVS CNV caller now overwrites older versions of reference samples that already exist (rather than erroring out).
- The Sample Summary Table spreadsheet from SVS CNV now shows the Percent Difference field as a true percentage.
- Logging out of SVS now clears information about the current licensed machine.
- The license flags are now kept when running .tsf convert, including from the stand-alone GenomeBrowse.
- SVS installations on servers that generate a different machine ID every time a user connects to the server now work correctly.
- The bundled unzip library for Linux has been updated to work with SVS marker maps.
- For Genotype > Mixed Linear Model Analysis and Genotype > Mixed Linear Model Analysis with Interactions, real or double precision data will now work when the Dominant or Recessive model is selected. This will allow taking Dominant-model or Recessive-model data, PCA-correcting it, then using those results as input to either of these Mixed-Model features.
- When adding a plot item to a GenomeBrowse plot from the current SVS project, the project view now always shows the spreadsheet column selection window on the right. Previously, that window would sometimes be absent.
- When adding a plot item to a GenomeBrowse plot from a different freshly-generated spreadsheet (that is in the current SVS project), the plot item will always appear and use the proper scaling (possibly after a fresh computation of mapping and scaling parameters takes place). Previously, sometimes a new plot item would either not appear or be of an incomplete scaling that did not cover its range properly.
- An occasional hanging up when exporting from SVS on Linux has been fixed.
- The following X-Chromosome-related issues have been fixed in the Genotype > Compute Genomic BLUP (GBLUP) and Genotype > Quality Assurance and Utilities > GBLUP Genomic Relationship Matrix features:
- For Overall Normalization, female X-Chromosome data is now more realistically scaled with respect to male X-Chromosome data and with respect to male and female non-X-Chromosome data.
- X-Chromosome data is now normalized and centered using the overall major and minor allele frequencies, rather than by separate male and female allele frequencies.
- Allele substitution effects (ASE) that are computed for the X-Chromosome now use data from both genders. Before, male ASE values were computed only with male data, and female ASE values were computed only with female data.
- Other issues fixed in the Genotype > Compute Genomic BLUP (GBLUP) and Genotype > Quality Assurance and Utilities > GBLUP Genomic Relationship Matrix features are:
- Normalizing a GBLUP Genomic Relationship Matrix using Individual Normalization now accounts for the monomorphic markers that were scanned.
- When a separate GRM is used for the X Chromosome, the allele substitution effect (ASE) for any marker is now calculated based only on which GRM to which that marker contributed. Thus, there will be only one pair of ASE value columns now output for this use case.
- Since by-marker effects calculated based on gene-by-environment GRM matrices are not really allele substitution effects, as such, SVS no longer uses this label for these values. (The label “Allele Activity” is used instead.)
- Some internal code changes were made that will eliminate “Future warning” messages from being written to the Python shell window.
- The following issues have been fixed in the Genotype > Predict Phenotypes from Existing Results feature:
- This feature now uses the correct allele substitution effect (ASE) data as input. Before, the first 350 ASE results were used in a repeating loop, and the skipping of monomorphic markers was inconsistent.
- This feature will now work even if a fixed-effect coefficient is exactly zero. Before, this situation would cause this feature to error out with a spurious error message.
- This feature will no longer crash if Correct For Gender is selected.
- The following issues have been fixed in the Genotype > K-Fold Cross Validation (for Genomic Prediction) feature:
- Selecting Correct For Gender no longer crashes the script.
- The Area Under the Curve test has been corrected. It used to show values that were sometimes very slightly above one.
- The genotypic and error variances for the GBLUP estimate spreadsheets are now displayed in their node change logs.
- For Genotype > Mixed Linear Model Analysis and Genotype > Mixed Linear Model Analysis with Interactions, canceling from Linear regression (fixed effects only) no longer crashes the script.
- The add-on script for converting binary and integer values to genotypes now applies the marker map correctly, even if there are unmapped fields in the spreadsheet.
- SVS annotations for categorical arrays are now correctly encoded in SVS spreadsheets.
- Building reference panels for BEAGLE imputation can now handle larger datasets without crashing.
- Annotation with CADD and annotating CNV’s have both been fixed to not crash SVS.
- File > Create Marker Map from dbSNP no longer crashes if there are no matching rsIDs.
- DNA-Seq > Variant Binning by Frequency Source has been fixed to accommodate an empty alternate frequency list in the Allele Frequency Annotation track.
- All of the DNA-Seq > Collapsing Methods > … options have been updated to accommodate transcript lists in the newer versions of the RefSeq track.
- The progress bar for DNA-Seq > Collapsing Methods > Count Variants per Gene now shows progress realistically.
- The progress bar for Numeric > Fishers Exact Test for Binary Predictors now shows progress more smoothly.
- Unnecessary dividing by zero in DNA-Seq > Collapsing Methods > SKAT-O has been removed. The main effect of this change is to clean up extraneous messages from being shown in the Python shell window.
- For Import > Third Party (from the project view), the nonsense file type “0 (*)” has been removed.
- The screen layout of DNA-Seq > Collapsing Methods > Mixed-Model KBAC has been improved to work properly under Linux.