In order to thoroughly assess a variant’s pathogenicity, it is important to take into account the variant’s effect on splicing. While the interpretation of variants that disrupt the pairs of bases at the beginning of a splice site is fairly straightforward, variants resulting in the introduction of a novel splice site are more difficult to interpret. In this blog post, we will discuss VarSeq’s capabilities for the interpretation of novel splice site variants, including a deep dive into the process of reviewing the clinical literature associated with such variants.
Each intron of a gene has two distinct nucleotides at either end which define the associated splice sites. The splice site at the 5’ end is called the donor splice site and is denoted by a GT nucleotide pair, while the splice site at the 3’ end is called the acceptor splice site and is denoted by an AG. The distribution of bases surrounding these nucleotide pairs is called the splice motif. Generally, methods for the detection of splice-altering variants rely on machine learning and probabilistic models which are used to identify the presence of these splice motifs.
Splice Site Prediction in VarSeq
VarSeq currently uses four algorithms to evaluate the effect of each variant on splicing:
- GeneSplicer: Uses Markov models in combination with maximal dependence decomposition
- MaxEntScan: Approximates sequence motifs using Maximum Entropy Distribution
- NNSplice: Identifies splice sites using neural networks
- PWM: Uses a position weight matrix similar to SpliceSiteFinder and Human Splice Finder
These algorithms are used to detect both the introduction of novel splice sites and the disruption of existing splice sites. For each of these algorithms, we provide a score, indicating the confidence of each algorithm on the presence of the nearest splice site, along with a prediction of whether the algorithm predicts the variant to be disruptive. VSClinical also provides detailed splice site predictions along with intuitive visualizations of each variant’s effect on splicing as predicted by each algorithm. This allows users to easily identify the introduction of novel splice sites, as well as disruptions of existing splice sites.
Evaluating a Novel Splice Variant in VSClinical
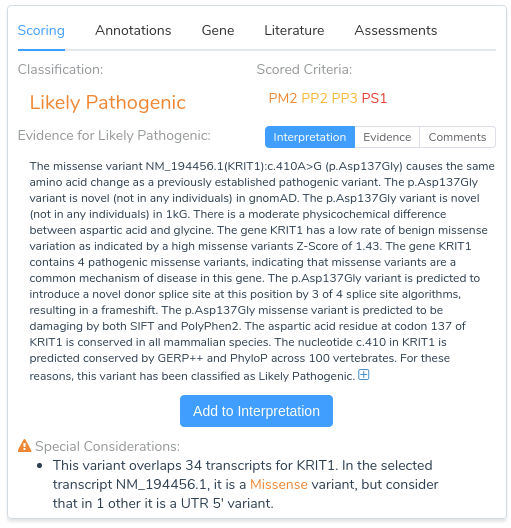
Now that we have provided a brief overview of VarSeq’s splice site analysis capabilities, let’s take a look at an example to illustrate the interpretation of a novel splice variant in VSClinical. Here we will be evaluating the missense mutation KRIT1 c.410A>G using the ACMG Guidelines workflow. Looking at the recommendations, we can see that there are three applicable criteria: PM2, PP2, PP3, and PP5.
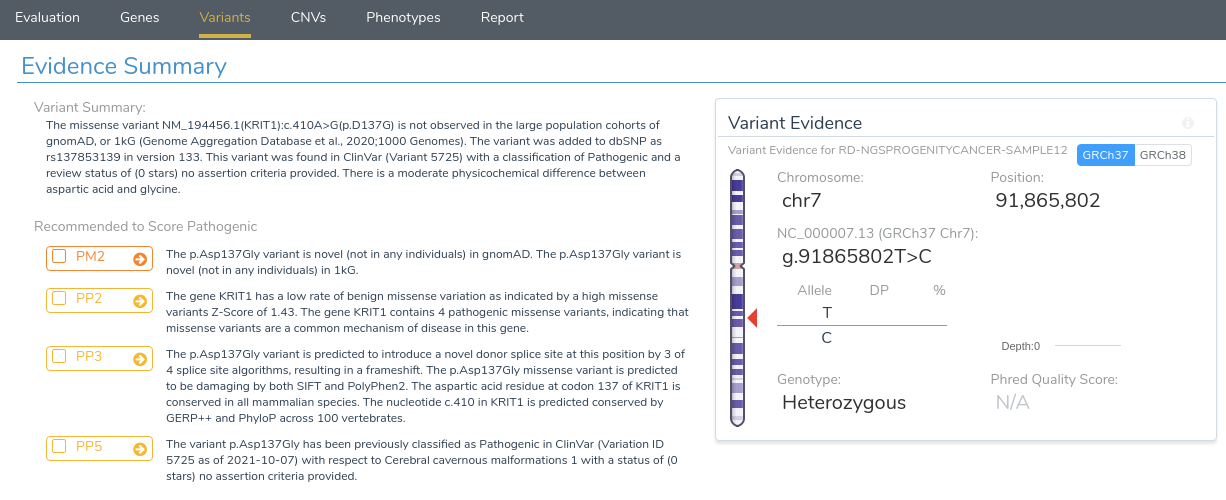
The first two criteria recommendations are fairly straightforward. PM2 is being recommended because this variant is absent from both of our population frequency catalogs: gnomAD Exomes and 1000 Genomes. PP2 is recommended because the gene KRIT1 has a low rate of benign missense variation and contains 4 previously classified pathogenic missense variants, indicating that missense variants are a common mechanism of disease in the gene.
Things get more interesting when we begin looking at the recommendation summary for PP3. Here we can see that the variant is predicted to introduce a novel donor splice site, resulting in a frameshift. Let’s take a closer look at the details for this recommendation by clicking on the arrow next to this criterion.
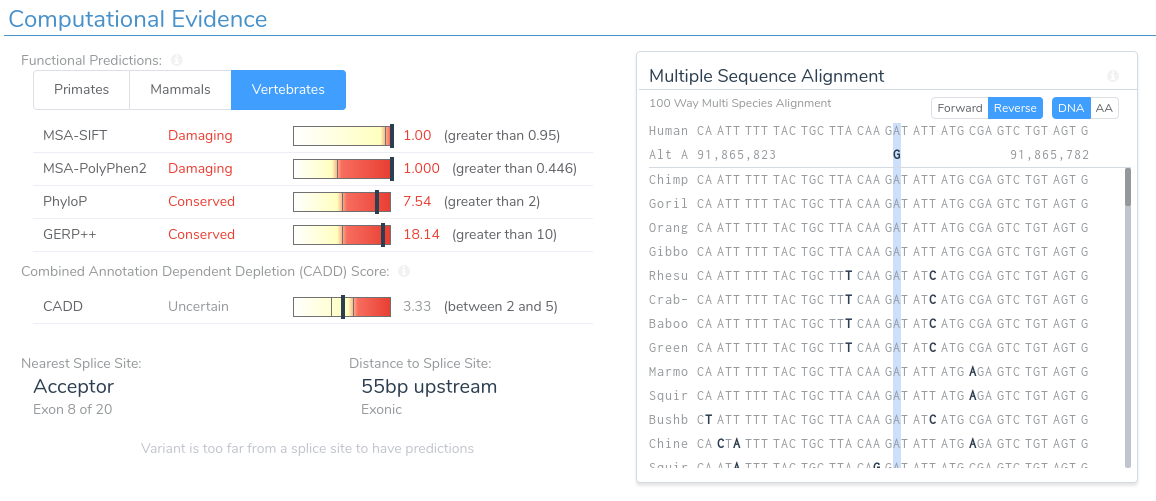
Here we can see that this variant is predicted as damaging by both SIFT and PolyPhen2. Additionally, the region is highly conserved according to PhyloP and GERP++. This can be verified by looking at the multi-species alignment chart, which shows that this nucleotide is conserved across all species in the alignment. If we scroll down, we can examine the splice prediction evidence for this variant.

Here we can see that the variant is predicted to introduce a novel splice site by three of the four splice site prediction algorithms. Additionally, VSClinical predicts that the introduction of a novel splice site at this location will result in a frameshift. If we open our splice site analysis visualization for this region we can get a more detailed picture of the variant’s effect on splicing.
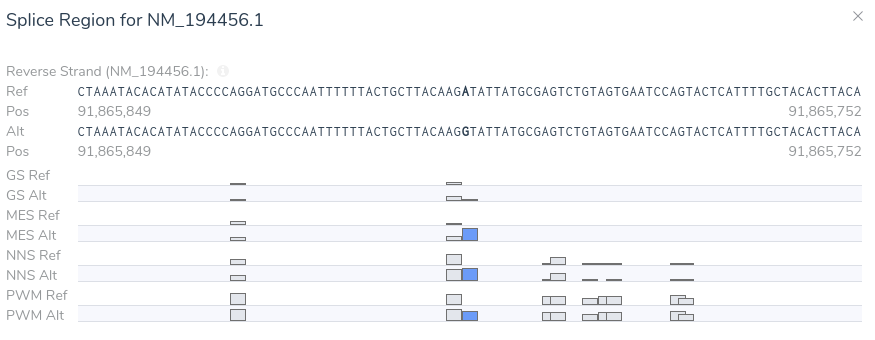
From this visualization, we can see that this variant results in an A > G mutation causing the introduction of a donor motif at this location. Additionally, we can see that all of our splice prediction algorithms have an elevated score at this position in the alternate sequence. Based on the above evidence, we can be confident in our application of criterion PP3, as all computational evidence supports a deleterious effect on the gene. Furthermore, the novel splice predictions provided by VSClinical motivate further investigation into the clinical literature to affirm our suspicion that this variant introduces a novel splice site.
Returning to our evidence summary, we see that criterion PP5 is recommended, as this variant has been previously classified as Pathogenic in ClinVar with zero stars. However, it should be noted that this criterion should only be applied as a last resort after failing to find additional evidence through a review of the existing literature. With that in mind, we will scroll down to the Clinical and Functional Studies section to review the literature associated with this variant. Here we can see a detailed assessment of this variant in ClinVar, along with associated publications in the clinical literature.

Looking at the publications associated with this assessment, we notice a paper describing missense mutations in KRIT1 leading to splicing errors in cerebral cavernous malformation. By clicking on this publication, we can see more details about the paper including the complete text of the abstract.
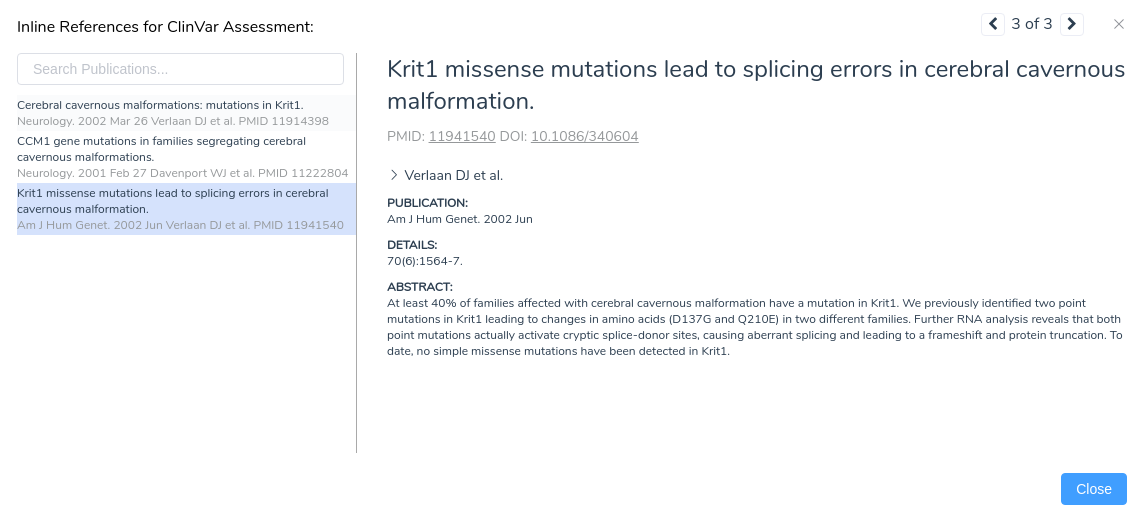
From the abstract, we can see that this paper describes the same mutation under evaluation in our project. Furthermore, the paper states that RNA analysis was performed on this variant establishing that the mutation activates a cryptic splice donor site, resulting in aberrant splicing and leading to a frameshift mutation causing protein truncation. With this additional evidence, we can confidently apply the criterion PS1, since the variant has been previously classified as pathogenic by a different laboratory with clear evidence supporting a deleterious effect through the introduction of a novel splice site. With the application of PS1, we now have sufficient evidence to classify the variant as likely pathogenic.
Conclusion
I hope that this blog post has provided valuable insight on how VSClinical can be used for the evaluation of novel splice site variants and their associated clinical literature. If you want to know more about our splice site analysis capabilities or any other features in VarSeq, please contact us at info@goldenhelix.com. Feel free to also check out some of our other blogs that always contain important news and updates for the next-gen sequencing community.