When the new human reference genome was released over two years ago, it was hailed as a significant step forward for next generation sequencing. Compared to GRCh37, the new GRCH38 reference assembly fixed gaps, repaired incorrect sequences and offered access to sections of the genome that had been previously unaccounted for. Despite these improvements, adoption of the new assembly has proven to be a headache for many researchers. Whether due to the lack of required annotations, or the weight of their archived data on GRCh37, some organizations have chosen to simply stick with the old assembly while others have begun using GRCh38 on new projects leading to data that can not be compared between the different versions.
Ultimately, organizations need some way to convert between assemblies in order to perform data integration, annotation and visualization. Two existing options for performing this conversion are UCSC’s Liftover and NCBI’s Remap tool. The first maps genomic positions between assemblies and performs strand-flipping where necessary, while the second converts genomic positions and performs more complex operations to convert the variant alleles. Both of these tools are freely available online, and can convert between a number of different assemblies.
At Golden Helix, we have developed our own implementation of Liftover and have made a few improvements to the algorithm. When a genomic coordinate is converted using Liftover, it is possible that the original reference sequence will not match the reference at the newly mapped position. One reason for this is that a variant that was considered an alternate in the source assembly is now considered the reference in the target assembly. An example of this can be seen by looking at rs1763642. In GRCh37, this variant had a Ref/Alt of “C/T” as shown below:
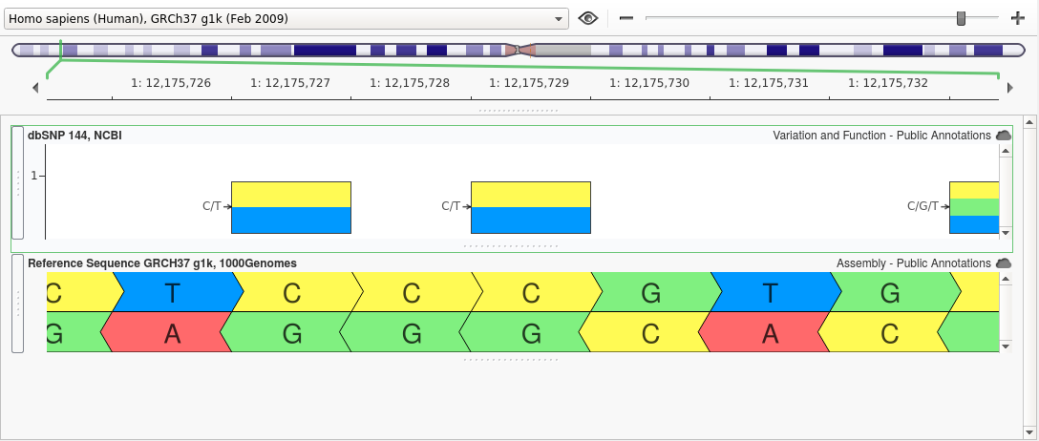
However, in GRCh38 the allele at this position in the reference assembly was changed from a “C” to a “T” and, as a result, the reference and alternate for rs1763642 were swapped as shown below:
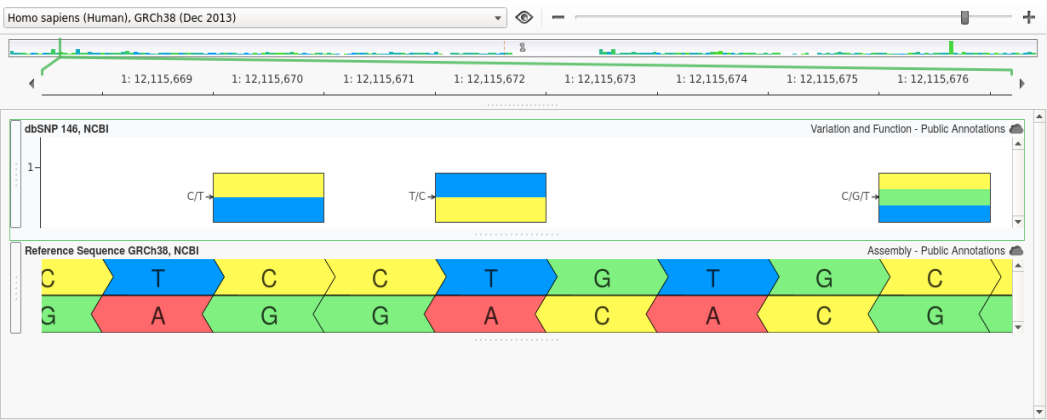
Our approach identifies these Ref/Alt swaps and corrects for them upon conversion by comparing the reference and alternate in the unconverted variant against the reference for the target assembly. If the reference doesn’t match but the alternate does, then we know that the Ref/Alt should be swapped when converting to the new assembly.
So, how does our implementation of Liftover compare to UCSC’s Liftover and NCBI’s Remap? To answer this question, we have run all three tools on a complete exome. We compared the percentage of variants that were successfully mapped from GRCh37 to GRCh38 using dbSNP coordinates and alleles on each assembly as a ground truth. For each algorithm, the graph below shows the percentage of variants that were correctly mapped to the corresponding dbSNP variant after conversion.
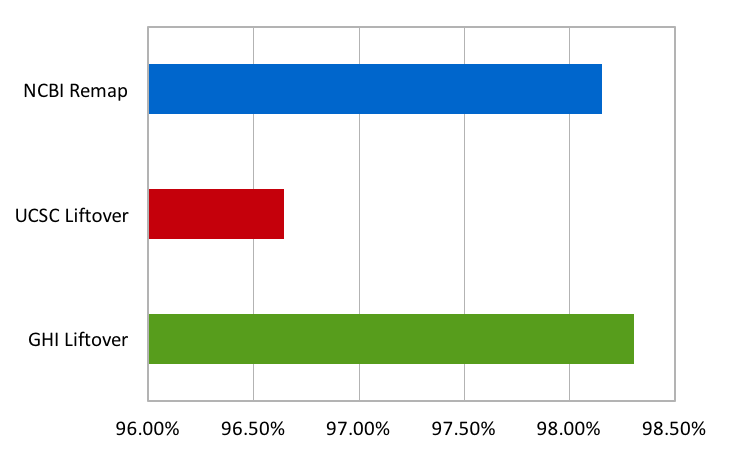
All three algorithms perform very well on this exome, successfully converting over 96% of the variants. Our implementation of Liftover has about a 2% improvement over USCS’s Liftover and has a success rate on par with NCBI’s Remap at around 98%.
Of course, there is more to the performance of these algorithms than the success rate. If an algorithm is unable to accurately convert a variant then, ideally, no result should be returned for that variant. Of the 35,718 variants evaluated above, NCBI’s Remap returned no results for 92 variants, however 568 of the converted variants failed to match the corresponding variant in dbSNP. For these cases, it would be better to return no result at all. In contrast, while our implementation failed to convert 484 variants, only 121 variants did not match their counterpart in dbSNP. One reason for this improvement is that we compare each converted variant to the target assembly and, if a mismatch between the reference sequence is found, then no result is returned.
Ultimately, the only way to get perfect conversion between two assemblies is to realign the sequence reads of your old data against the new reference assembly and for most labs, this is simply not an option. While none of these methods are perfect, all of the algorithms discussed here will correctly convert the vast majority of variants.
We are currently in the process of incorporating our implementation of Liftover into VarSeq and soon you will be able to utilize this powerful tool on your own data!
Hi, I was wondering about variants that occur between GRCh37 and GRCh38 but not between GRCh37 and the genome to be remapped- wouldn’t those need to be incorporated into the new file also? Say that in one region, both assemblies are identical except for one base, where GRCh37 has an A and GRCh38 has a G. The original vcf file has no variant at that base, so the genome to be remapped also had an A at that base. But once you map the coordinates, wouldn’t it now need a SNP at that location, where Ref is G and Alt is A?
Your are correct. In the case you describe, a variant that was not present based on the original assembly would now be present according to the new assembly. Based on this scenario, if such a variant was present in the vcf at that position in GRCh37 then it would be called as A/G with a genotype of 0/0. Since the reference at that position in GRCh38 is now G, that variant would be flipped so that it becomes G/A. However, if there was no variant at that position in the original vcf then no variant will be reported at that position after the conversion.