
VSClinical AMP Matching of Interpretations
In this blog post, we will delve into the intricacies of the VSClinical AMP interpretation workflow. At the heart of this process lies the task of annotating cancer biomarkers with the correct interpretations based on the classification of the tumor and the type and scope of the biomarker. This is a crucial step in understanding the patient’s cancer and determining the best course of treatment. We will explore how the AMP Guidelines determine the best interpretations to add and report based on the genomic data available.
AMP Interpretation Workflow: Annotating Cancer Biomarkers
At the core of the VSClinical AMP interpretation workflow is annotating variants, CNVs, fusions, and genomic signatures with the interpretations that match the patient’s tumor type and the biomarkers in the evaluation. This differs from variant annotation, where the exact variant can be saved and retrieved from a public annotation source or a catalog of previously seen variants. The AMP Guidelines provide a method to assess the strength and tier of evidence for drug sensitivity, drug resistance, and diagnostic and prognostic reportable data. To make the evidence something that can be saved and applied to future samples, the correct biomarker scope must be determined. Does this drug label indication apply to all activating variants in a gene? Maybe all EGFR exon 19 in-frame deletions? Or has it been shown to be relevant to all BRAF V600 codon mutations?
Let’s first look at how VSClinical supports saving and applying interpretations with flexible biomarker scopes. This enables customizing the automated interpretation workflow of VSClinical AMP to match the focus and curation work of your NGS test.
Evaluation Biomarkers
The starting point of the interpretation lookups is the biomarkers added to an evaluation and the tumor type of the current sample. VSClinical automatically matches interpretations from both the configured internal Knowledgebase as well as from CancerKB. It does this whenever a biomarker is added, removed, or modified. Along with the patient’s tumor type, the following properties of a biomarker are used to lookup and match interpretations:
- Gene: Interpretations only match for interpretations that reference the same gene or genomic signature
- Region: Interpretations can be written for a specific region of the gene and will only match if that region overlaps the biomarker’s region
- Scope/Type: (i.e., Amplification, Missense) Biomarkers will only match interpretations written for a matching or compatible scope
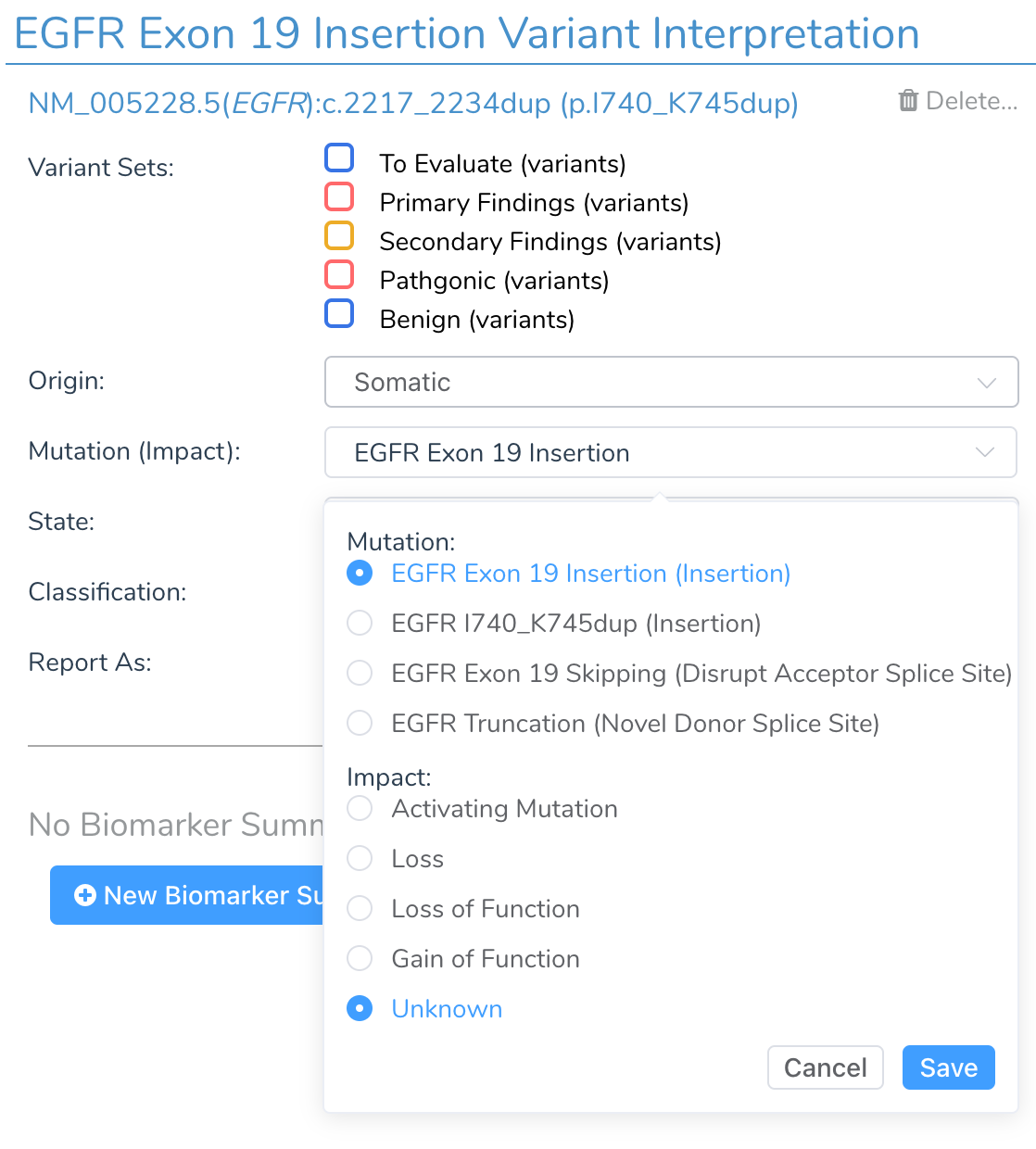
- Impact: (i.e., Activating, Loss of Function) Interpretations that are written for genes, exons or codons instead of variants will specify an impact, which must match that of the biomarker to apply the interpretation. (I.e., BRAF activating mutations)
When a variant is added to VSClinical, it may have an impact automatically assigned. If the variant has an auto-classification or previously saved classification of Oncogenic, then the impact will be set to Activating for variants in oncogenes. Similarly, if the variant is set to Oncogenic and is a truncating mutation (i.e., a frameshift insertion), then the impact will be automatically assigned to Loss of Function when in a tumor suppressor gene. A variant can also have the impact set when from a matching biomarker summary interpretation, allowing the CancerKB and internal Knowledgebase to curate common mutations that have known impacts.
Interpretations and their Biomarker Scopes
To match evaluation biomarkers, cancer interpretations are written for biomarker scopes. Interpretations can have one or more “biomarker scopes” specified. Biomarker scopes can be very specific, matching only a single variant, or very broad, matching every variant for a gene. Outside of matching a single variant, it can be written for ranges of codons, exons, or the entire gene. When a biomarker scope is defined for a region, it must specify the type of variants in the regions it should match. For example, truncating mutations, missing variants, or all variants. Finally, interpretations that specify an AMP tier should define a biomarker impact so that variants that don’t meet the functional or scoring threshold and have an unknown impact do not match the interpretation.
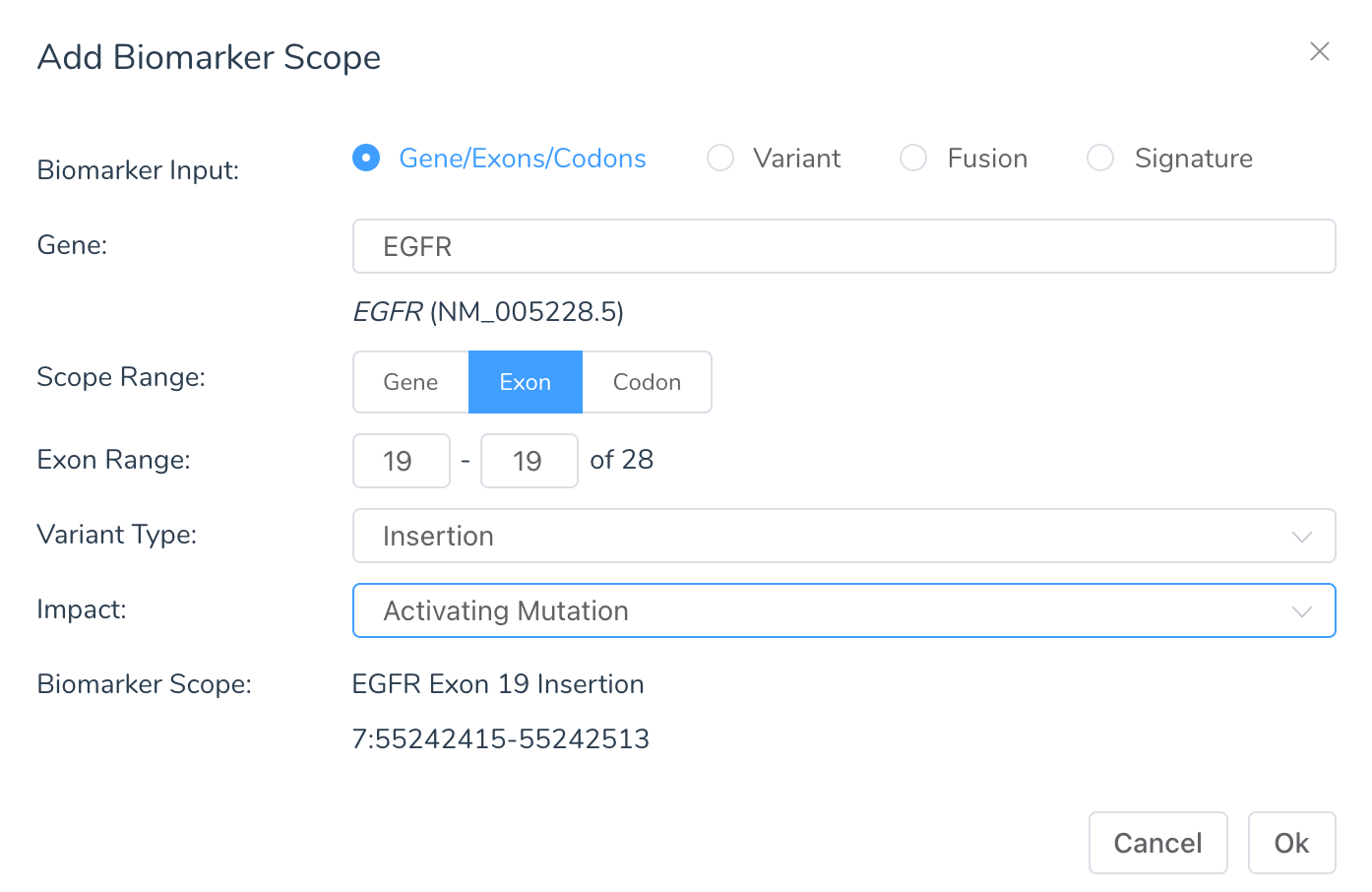
When creating a biomarker scope, there are four input types:
- Gene
- Variant
- Fusion
- Signatures
When using the gene input, the biomarker scope will be a region defined by a range of codons, exons or the whole gene with a specific scope and impact. The following variant types can be specified:
- Mutation: Most generic, will match any biomarker in the gene
- Amplification: Match CNV duplications
- Deletion: This can be both whole gene deletions and in-frame partial deletions of the gene. For example, in-frame deletion variants (one or more codons), exon skipping splice variants, and in-frame exon deletions from CNVs.
- Insertion: In-frame insertion variants (one or more codons) as well as in-frame exon duplications from CNVs
- Truncation: Any mutation that disrupts the reading frame or introduces an early stop. For example, nonsense, frameshift indels, exon-level CNVs that are not in-frame, and splice variants that are not exon skipping
- Nonsense: Specifically, nonsense (stop-gain) variants
- Splice Mutation: Specifically, variants that mutate the canonical splice regions
- Skipping: Loss of in-frame exons by mutating the acceptor splice site
- Missense: Variants introducing an amino acid change
- Noncoding Mutation: Variants in non-coding transcripts or UTR regions
- Silent Mutation: Synonymous variants and stop retained variants
With that in mind, let’s dive deeper into the concept of variants and their scopes.
Variants and their Biomarker Scopes
When variants are added to an evaluation, they are assigned a list of possible scopes based on their sequence ontology, position in the gene, and predicted impact. For most cases, the variant’s default scope will already be set up to match common interpretations.
In some cases, how a variant is thought to act at a functional level will change what type of interpretations it should match. For example, a synonymous variant at the end of an exon boundary will, by default, match biomarker scopes for a “Silent Mutation” or “Mutation.” But if the variant is suspected to impact the adjacent donor splice site, then the type can be changed, and the variant will be interpreted as a truncating splice mutation and match interpretations for that type.
VSClinical automates this change when there is strong in-silico splice site predictions that indicate that a variant is functionally splice disrupting. If extra functional evidence is available one way or another, the user can change the variant type manually.

Small inframe insertion and deletion variants in cancer are often activating mutations and treated interchangeably at the exon level. For this reason, inframe indels automatically have their mutation type default to be reported as exon-level insertions or deletions. For example, the NM_005228.5(EGFR):c.2217_2234dup(p.I740_K745dup) variant in EGFR will be displayed as Exon 19 Insertion and the NM_005228.5(EGFR):c.2238_2255delinsGCAACA(p.L747_S752delinsQH) variant as an Exon 19 Deletion. The variant can be switched to being treated as a specific variant and not an exon-level variant at the user’s discretion.
Flexibility and Automation for Cancer Workflows
With the improvements to VSClinical AMP in VarSeq 2.3.0, the user can precisely define the genomic biomarkers or multiple biomarkers that should match a reportable cancer therapeutic or diagnostic finding. With the starting point of Golden Helix CancerKB and the power of the biomarker scope matching system, labs can streamline the complex process of reporting out samples with precision medicine insights that are actionability for the treating physician and oncologist.