VSClinical is our most recent product that allows users to evaluate variants according to the ACMG guidelines. As with any tertiary analysis, there is a need to implement best practices into your workflow and using VSClinical for the ACMG guidelines is no exception. That said, we have put together a Best Practices Blog Series, with the purpose of discussing some essential elements of VSClinical that users can account for when integrating this powerful interpretation tool. This article will focus on implementing the ACMG classifier into your filter chain to capture clinically relevant variants that might otherwise be filtered using a standard workflow.
As you may know, the number of variants in a VCF file depends on the initial target region, which can range from hundreds of thousands to millions of variants. Filtering is performed to reduce the number of variants to those that have high clinical relevance, are high quality, have rare frequencies or are novel, and are predicted functionally damaging. As an example, a primary filter chain for a targeted gene panel may consist of filtering variants for sufficient read depth and genotype qualities as well as minor allele frequencies to identify rare variants.
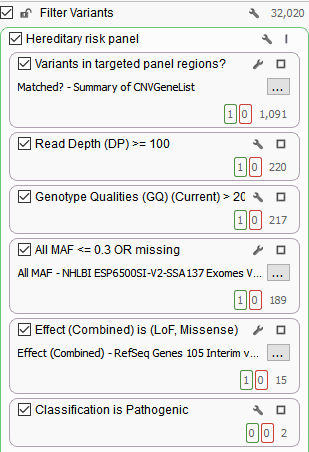
In addition, this filter chain is also filtering on Effect (Combined) from RefSeq Genes and selecting for pathogenic variants according to ClinVar. As mentioned in a previous blog, the Effect (Combined) specifically targets Loss of Function and missense mutations. Utilizing these annotations as filters significantly reduced the total number of variants from 32,020 to two variants that are clinically relevant. Although these variants are associated with pathogenic classifications, it is also important to not ignore interesting synonymous variants.
Synonymous variants do not alter the amino acid sequence of the protein and are thought to be silent mutations. However, recent literature has highlighted that synonymous variants can affect protein splicing, expression, and function that ultimately can contribute to disease. Thus, we propose an alternative to using the Effect (Combined), which filters out synonymous variants. Our alternative is using the Classification field from the ACMG Classifier in your filter chain.
The Classification field leverages 18 of the 33 ACMG criteria to provide a classification for a variant but will also account for previous variant evaluations in VSClinical. This provides the first benefit, which is if you have previously classified a variant, the Classification field will alleviate you from having to re-evaluate the variant. The second attribute is that the Classification field is utilizing a variety of variant evidence, including population and functional data and computational prediction algorithms. This means that in contrast to prioritizing LOF or missense variants, as shown below, the Classification field will capture all clinically relevant variants, including those that are synonymous.
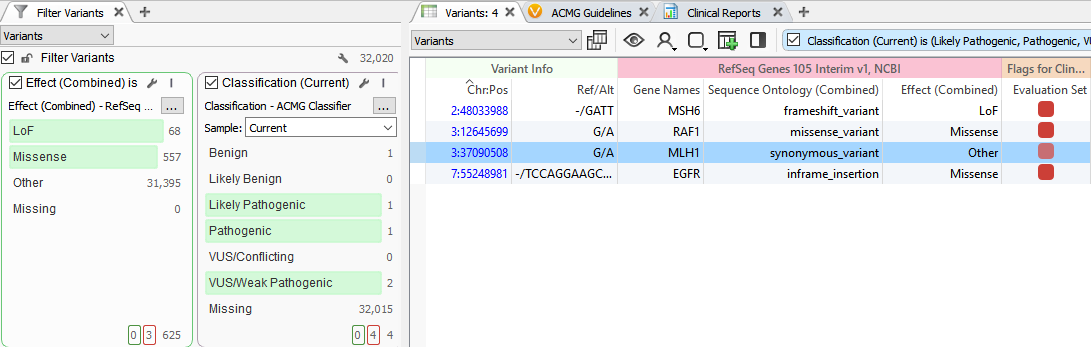
A prime example is a synonymous variant located in the MLH1 gene that was auto-classified as VUS/Weak Pathogenic. Filtering with the Classification field, this variant was captured and was then evaluated in VSClinical. Scoring this variant according to the ACMG guidelines resulted in a final classification of Likely Pathogenic as it influenced four splice site algorithms including Gene Splicer, MaxEntScan, NNSplice, and PMW. Additionally, there was also published literature citing this variant as Likely Pathogenic further supporting our final classification. Ultimately, this Likely Pathogenic variant associated with Muir-Torre Syndrome would have been lost if prioritizing missense/LOF variants alone.
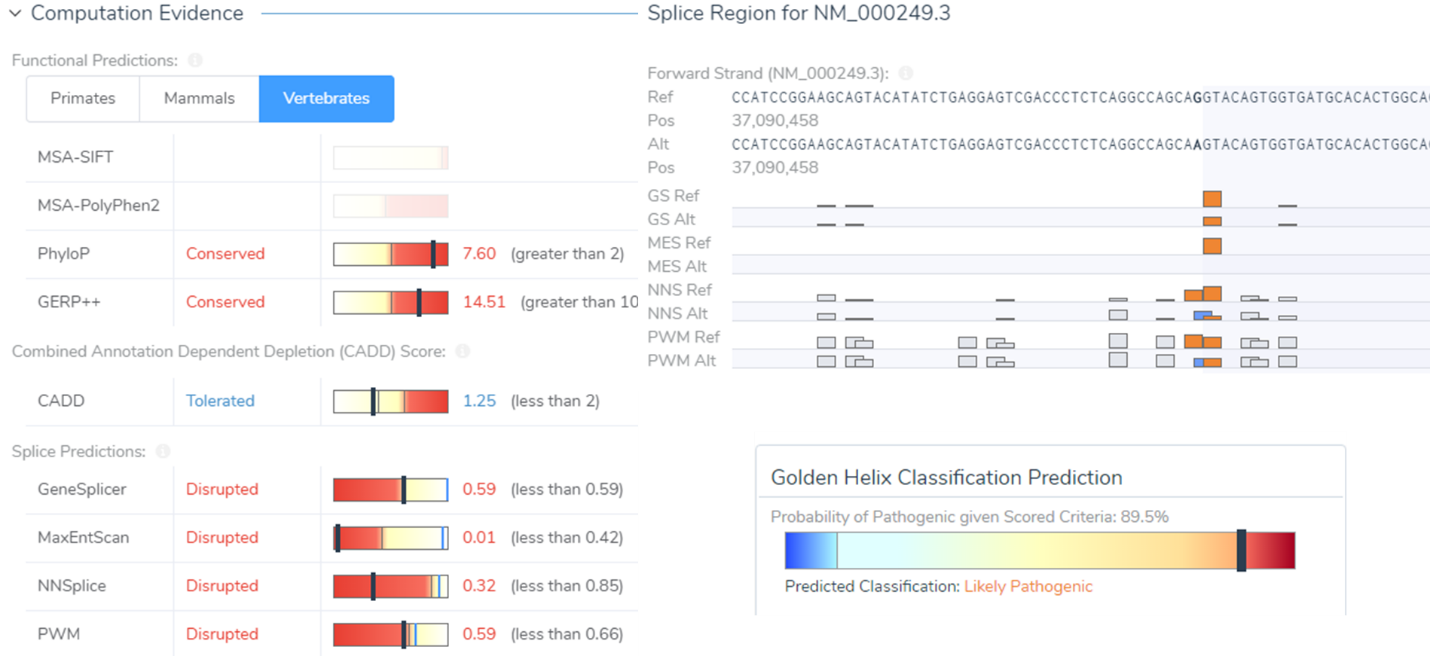
In line with capturing all clinically relevant variants using the Classification field, it is also worth mentioning the requirements for a variant to be evaluated with the ACMG guidelines. The first is that the variant needs to have sample level information and include a genotype field in the VCF file. Without these components, a variant cannot be evaluated in VSClinical unless manually added. The second is that the variant genotype should be a non-homozygous reference. In other words, heterozygous or homozygous variants are prime candidates for evaluation. Lastly, the variant must fall within a gene region. This is based on the classification approach, described by the ACMG Standards and Guidelines, which is intended for variants that fall within Mendelian genes.
Implementing the Classification field from the ACMG Classifier in your filter chain will ensure best practice of VSClinical. Utilizing this feature will not only capture all clinically relevant variants but will also help a user understand if the variant has been previously evaluated. This concludes the first blog of this series but if you would like a more fluid breakdown of this feature, please watch this great webcast or contact us at support@goldenhelix.com.
Continue this best practices workflow by checking out part II which focus on the caveats and interpretations of VSClinical! View Part II Here.