Smoothing Hurdles into Speed Bumps when creating Annotation Sources
Although most researchers assume that getting the pile of VCF sequence files is the largest hurdle in moving towards an analysis, there still exists the looming step of normalizing the variant calls in annotation sources to make variant comparison easier. In this ever-refining field of study, VarSeq continually works to increase the ease of allowing users to move from variant annotations to robust, normalized variant sets.
VarSeq Convert Wizard to the Rescue
This Convert Wizard allows users to convert files into easy-to-use annotation sources. Annotation sources can be associated with generic regions or specific variants. When converting a variant-based annotation source, you will want to consider the following normalization options:
- Distilling complex variants down into their allelic primitives by splitting MNPs into SNPs
- Left-aligning variants where needed to preserve canonical positioning
- Lifting over variants from one assembly to another
The first step in converting files into annotation sources in VarSeq is performed by importing the files into the Convert Wizard.
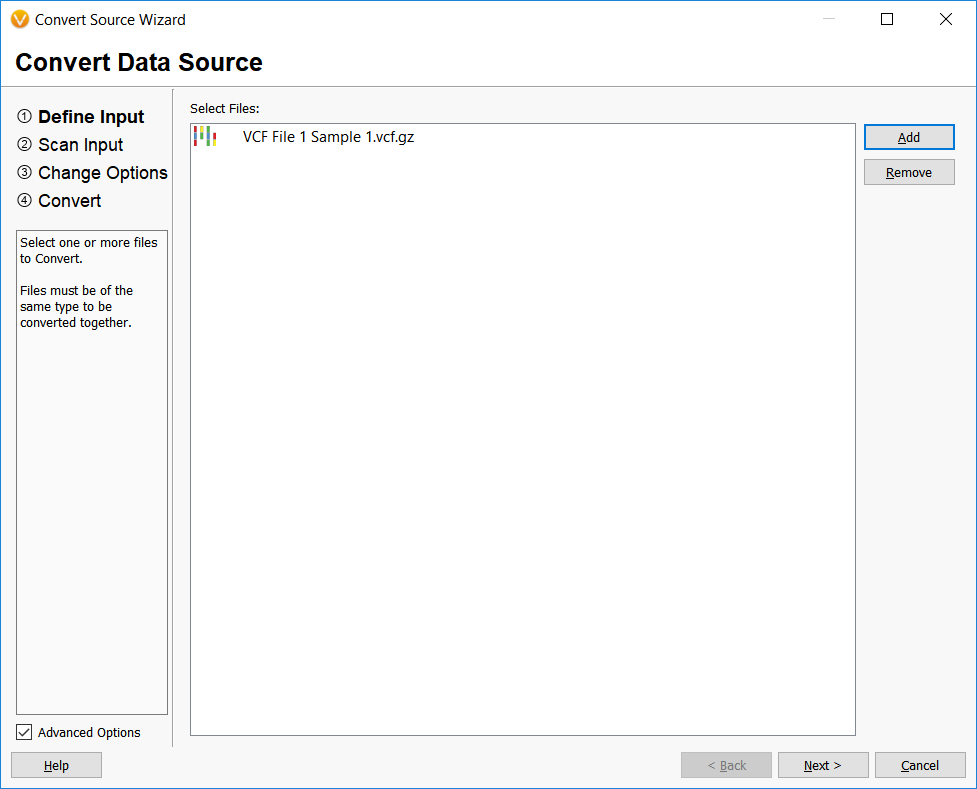
The input file assembly is matched and prompts allow the user to add any documentation or adjust field names.
The final step in the Convert Wizard is where the real normalization occurs. This step provides some additional options for the user to Left Align insertions and deletions, split multi-nucleotide polymorphisms into their SNP allelic primitives, and to Liftover to a different assembly if the annotation source and annotation target assemblies do not match.
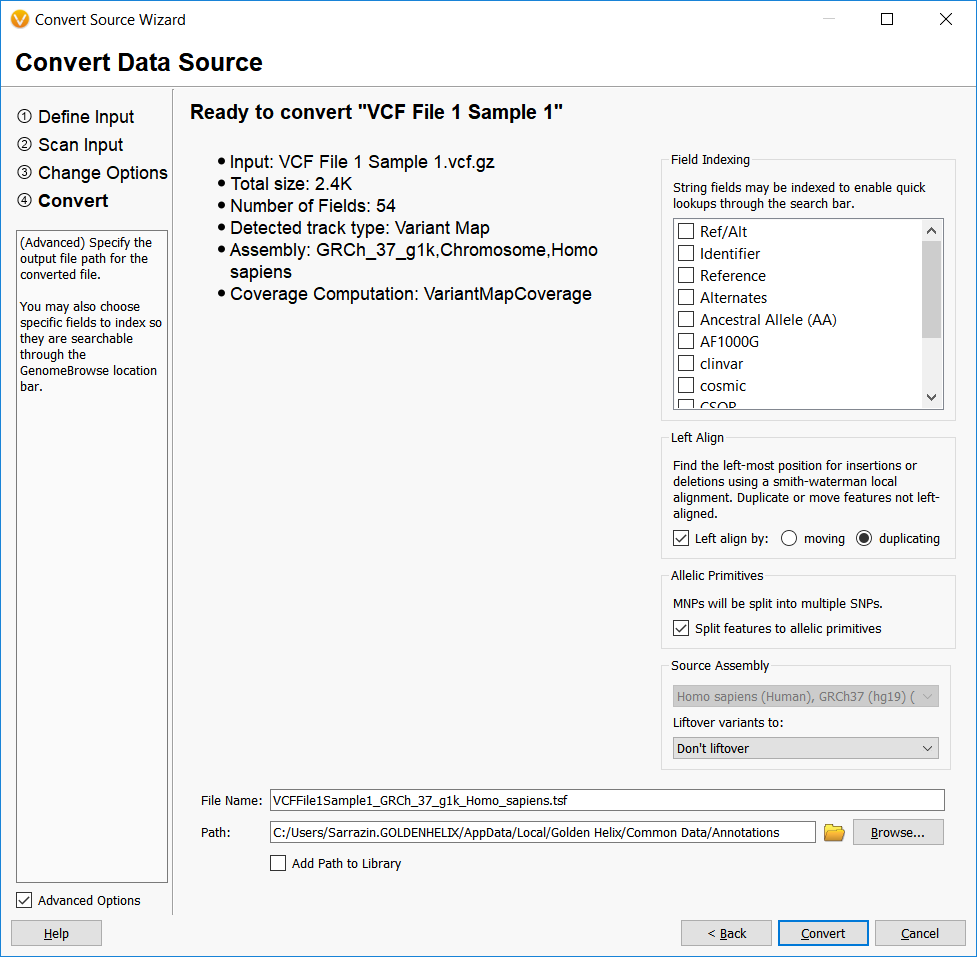
Before we further explore these options, it should be noted that all of these options stem from the main issue with variant normalization and that is what is the best way to represent an insertion or deletion?
Variant Normalization
To get to a single consistent representation of a variant, just follow 2 rules. A variant is normalized if (and only if) the variant is left aligned and parsimonious. Or put another way: a variant is normalized if it is described in its leftmost genomic position in the shortest format possible. A common example below shows the different ways a variant can be represented if the main 2 rules are not followed.
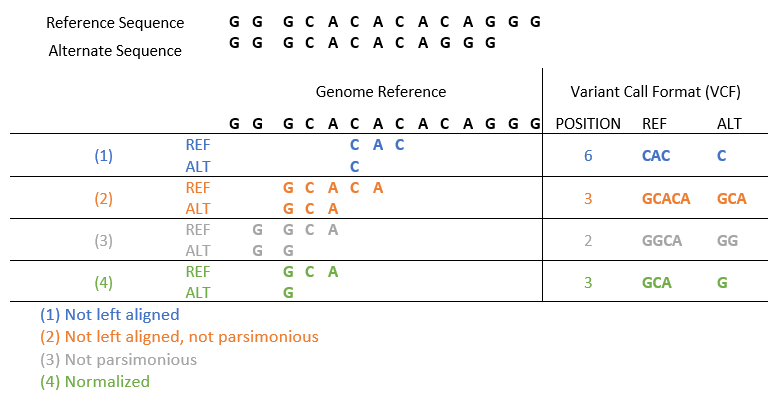
The first case does not have the variant left-aligned, so the middle ‘CAC’ sequence was selected as a deletion here, but not the leftmost, so the first rule of variant normalization is not met. The second case voids both rules since the ‘CA’ deletion is neither left-aligned nor parsimonious or described in its simplest form. The third case is left aligned but fails the parsimonious rule by not being displayed in its simplest form. And the fourth case finally satisfies both rules for the deletion.
Now, the Convert Wizard offers the option to left align, but also to perform the left alignment that moves the variant entry or perform the left alignment and keep the original entry as a duplicate. The example below shows the reference sequence on the top row, the original file on the second row, a left align with duplicate in the third row, and a left-align with the ‘moving’ option checked on the last row.
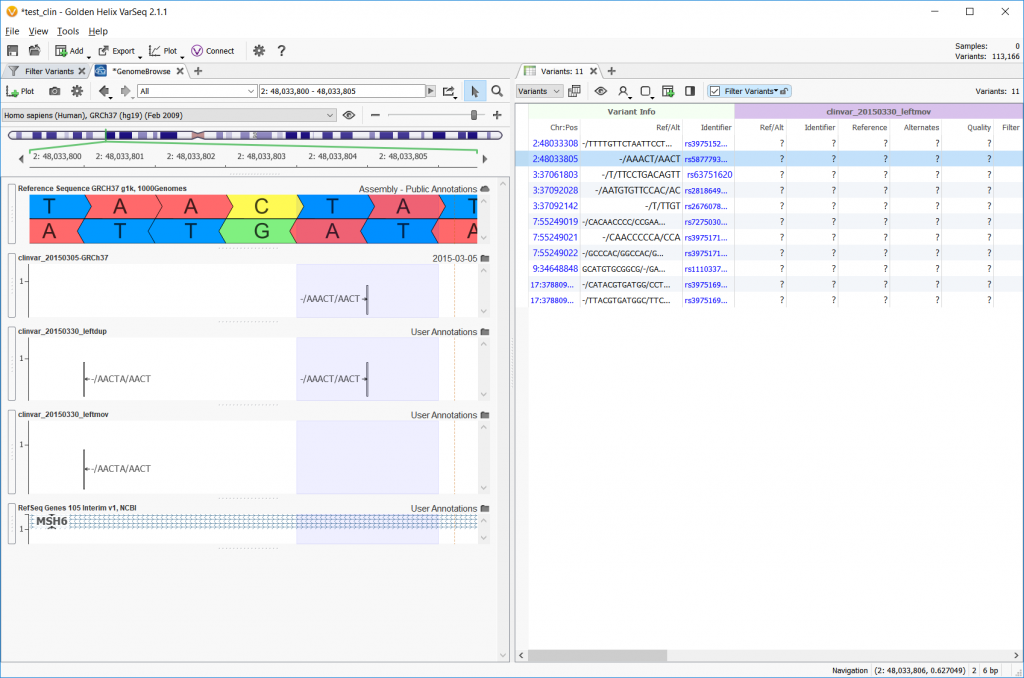
The next option in the Convert Wizard concerns differentiating multi-allelic variants and the allelic primitives that makeup multi-allelic variants. Checking this option and left aligning will split up any multi-allelic variants into the composite allelic primitives and possibly shift the SNPs to be left-aligned. The example below shows one example where an A/AA/- allele is split into its -/A and A/- allelic primitives.
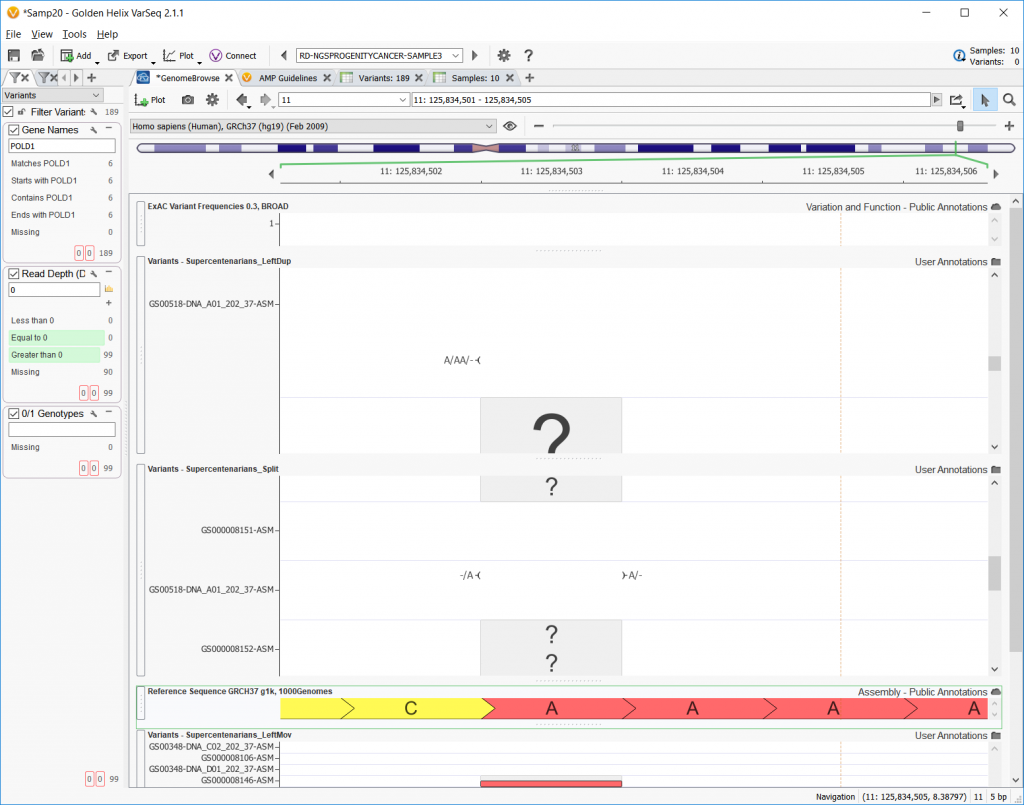
For more detail on this, please take a look at one of our previous blog posts on complex to primitive allele manipulation or variant normalization.
The final option simply allows users to liftover the assemblies of their annotation tracks to match their target data between GRCh37 and GRCh38. Some annotation sources are only supplying data in one assembly, so this tool makes it easy to adjust for use with different assemblies.
All of these tools combined will make incorporating different annotation sources much less of an obstacle and increase the accuracy of comparison.