I want to take this opportunity to highlight and briefly discuss some of the key features and updates that have been incorporated into VarSeq 2.2.3. Some of you may have attended the webcast that covered the prominent new features added to VarSeq, which are the updates to improve whole-exome analysis workflows, namely improved CNV calling in whole-exome datasets. However, there have been many other features added to VarSeq 2.2.3 that I think are worth emphasizing as well!
As I eluded to earlier, the front and center features for this release are the updates made for VarSeq-CNV. New filters and quality flags were incorporated into the CNV calling algorithms to ensure that high quality, true positive CNV events are being analyzed in whole-exome workflows. In the same vein, users can use the new “Create Low-Quality Target Regions” wizard to define a set of targets that tend to fail various quality metrics. An example of target regions included in this track are regions where the average depth across reference samples is shallow. Outside of optimizing the CNV caller for exome analysis, there were updates made to importing external CNVs into VarSeq, and CNVs can now be exported from VarSeq as VCF files.
There were also notable updates to VSClinical ACMG and AMP. Firstly, I want to mention that the splice site prediction algorithms used for both canonical and novel splice site detection are available to all VarSeq users (previously part of VSClinical)! If you have projects with gene annotations such as RefSeq Genes already incorporated, to enable splice predictions, you can right-click on the table header with the gene source name in the variants table and select Edit. The right-click option to edit algorithms is a new feature incorporated into VarSeq 2.2.3 and can be applied to any algorithm with user-defined input options. Figure 1 shows the right-click edit menu for opting in to the new transcript annotation options and splice site detection.
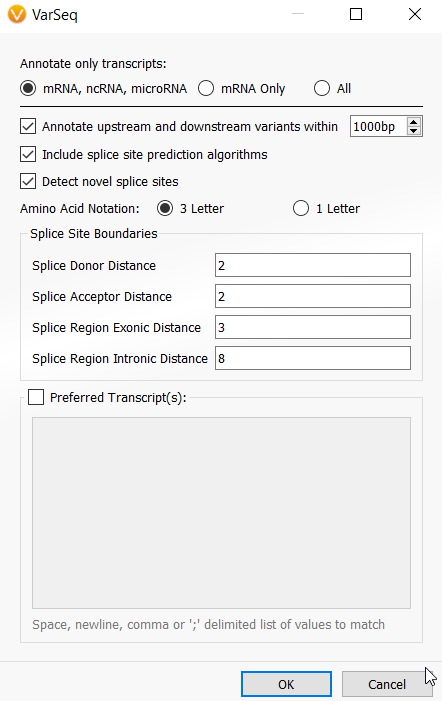
While I am discussing the transcript algorithm, note that there is now an option to annotate a subset of transcripts that includes non-coding RNAs and microRNAs in addition to mature RNAs (mRNAs) that was the previous default. Additionally, there is a new option to annotate variants that are upstream or downstream of interpreted transcripts. In addition to the VarSeq annotation algorithms, VSClinical will now support interpreting non-coding and up/downstream variants. In light of detecting these more unique variants and improvements to the splice site prediction algorithms, many of the recommended ACMG scoring criteria and strengths of the criteria have been adjusted. Many edge-case variants inspired these changes, so I would encourage exploring the VarSeq 2.2.3 release notes if you want more details on which criteria were adjusted and why.
On the VSClinical AMP side of things, the biggest update was to change the AMP interface to match the ACMG interface. This includes consolidating the first two tabs into an “Evaluation” tab and adding an expansive top menu available from any tab. Ultimately, this update helps improve the efficiency of AMP workflows with quick access actions to finalizing reports, close the evaluation and jump to the variants and biomarkers in the evaluation with a single click.
For all of the VSPipeline users out there, VSpipline saw many changes with VarSeq 2.2.3 that are certainly worth trying out with your projects! There are new commands available to allow the automation of different workflows, and overall there is better error message handling. One example of a new command added is liftover=true, which will allow users to use VSPipeline to import VCF files with different coordinate systems than the genome assembly specified in the project template. An example of better error handling that goes hand-in-hand with this capability is that if a VCF file does not match the genome assembly of the project template, an error will describe the mismatch.
I have only scraped the surface on the new features that are available in VarSeq 2.2.3. I would recommend browsing the VarSeq 2.2.3 release notes, specifically the section describing features added to the general projects and algorithm section. In addition, check out the blog post discussing the changes and updates to the gene preference systems! A complete list of features that have been added to VarSeq in this release can be found here. Please reach out to support@goldenhelix.com if you have questions about any new features or need assistance upgrading to VarSeq 2.2.3!