VarSeq 2.2.2 was released on December 17th, 2020 and the main feature that was added to VarSeq was that the VSClinical ACMG Guidelines workflow now has an additional CNV interpretation framework based on the ACMG/ClinGen guidelines. This product supports interpreting CNVs detected with VS-CNV or imported CNVs alongside variants and requires both a VSClinical ACMG license and a CNV license. Many other features have been incorporated into the release as well and so I want to take the opportunity to briefly touch on these additions.
Before diving in, I want to mention that we have demonstrated the application of the ACMG Guidelines/ process for scoring CNVs and discussed many of the new features in a series of webcasts.
- VSClinical: First Commercial Product to Integrate the Updated ACMG Guidelines for CNVs
- A User’s Perspective: ACMG Guidelines for CNVs in VSClinical
- Evaluating Copy Number Variants with VSClinical’s New ACMG Guideline Workflow
- Exploring New Features and Clinical Reports in the ACMG Guideline Workflow
VS-CNV: New Features and Polishes
Several new algorithms have been added to VarSeq- CNV and run on the CNV table:
Sample ACMG CNV Classifier: Much like the ACMG Sample/Variant Classifier algorithms, this algorithm computes classifications for each CNV based on the ACMG CNV guidelines.
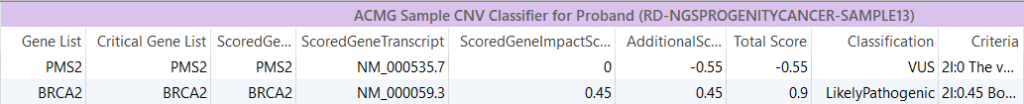
Copy Number Probability/Segregation: Computes the expected copy number of each called CNV. If parental information is provided for a sample, the algorithm also computes the probability that the CNV is present in the mother and the father. This is useful for having a computed confidence that a CNV is de Novo.

Latest CNV Sample Assessments: Annotates the CNV table with the latest assessments from a selected CNV assessment catalog.

Annotate CNVs Matching Current Sample: Annotate the CNV table with the assessments from a selected CNV assessment catalog that contain the current sample name in a specified “Samples” field.
Many existing CNV algorithms have been updated or enhanced:
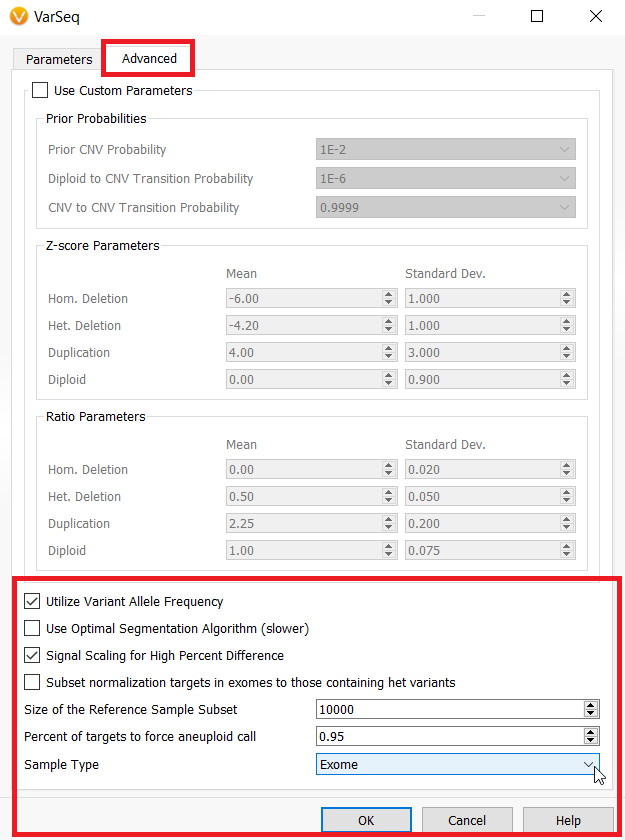
- CNV Caller on Target Regions. These changes can be found in the “Advanced Parameters” dialogue.
- The default value of “Size of the Reference Sample Subset” in the Advanced Parameters for the CNV Caller on Target Regions has been increased from 100 samples to 10,000 samples. Namely, this update can improve the normalization process in some cases.
- The checkbox “Signal Scaling for High Percent Difference” allows manual (enabling or) disabling normalization scaling performed on samples with a high percent difference to the reference samples.
- The new parameter “Percent of Targets to Force Aneuploid Call” forces an event to be called a whole chromosome event if the percentage of targets in a given state exceed the selected percentage (95% default). In other words, if the percentage of deleted or duplicated targets in a chromosome exceed the selected threshold, then the entire chromosome will be called as an aneuploid deletion or duplication event.
- The new parameter “Sample Type” can be set to either “Auto”, “Gene Panel”, or “Exome” for inferring the sample type. If the sample type is “Exome”, the algorithm is permitted to call whole chromosome events and report these events in the Karyotype output at the sample level as being whole chromosome events. You may force this option if you desire to call whole chromosome aneuploidy events, especially in the X or Y chromosomes.
- The option “Subset normalization targets in exomes to those containing het variants” will cause the algorithm to only use targets containing variants with a heterozygous VAF when computing the mean coverage for normalization. This is recommended only for highly mutated tumor samples. In previous releases this action always took place for “exome” sized target lists, but now must be turned on explicitly as an option as most samples do not benefit from this normalization technique
Adjustments and additions to the the CNV output
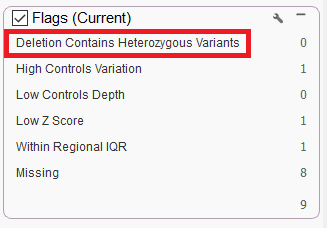
- The CNV output of a karyotype value has been adjusted to only report a karyotype on CNVs larger than at least one cytoband. CNV flags will also be reported for large CNVs along with karyotypes.
- A new flag was added to the CNV table output which denotes deletions that contain two or more heterozygous variants. This flag is called “Deletion Contains Heterozygous Variants”.
VSClinical ACMG: New Features and Polishes
The layout for the ACMG VSClinical interface has been updated:
- The existing ACMG variant scoring workflow has been reorganized into a single view under the Variants tab.
- The new ACMG CNV scoring workflow has been created under the CNV tab.
- The Gene tab now contains coverage statistics at the target level as well as the ability to provide a gene list for summary statistics and reporting.
- The Phenotype tab allows selecting phenotypes and disorders for the patient as well as entering patient notes that result in automatically extracted terms.
- The Reports tab summarizes all reportable information and the ability to render that information into a customizable Word and PDF report output.
The management of annotation sources used by the ACMG (and AMP) guidelines has been updated to allow selecting specific versions to be used in the evaluation process. The versions selected when creating an evaluation will be locked. This means that regardless of what new or updated sources are downloaded, an evaluation will continue to use and display the versions of sources selected on creation. The ACMG workflow allows these versions to be locked to the project template so all new evaluations are created with a specific version list. The name and version date of each source is available to display in the Word-based report system.
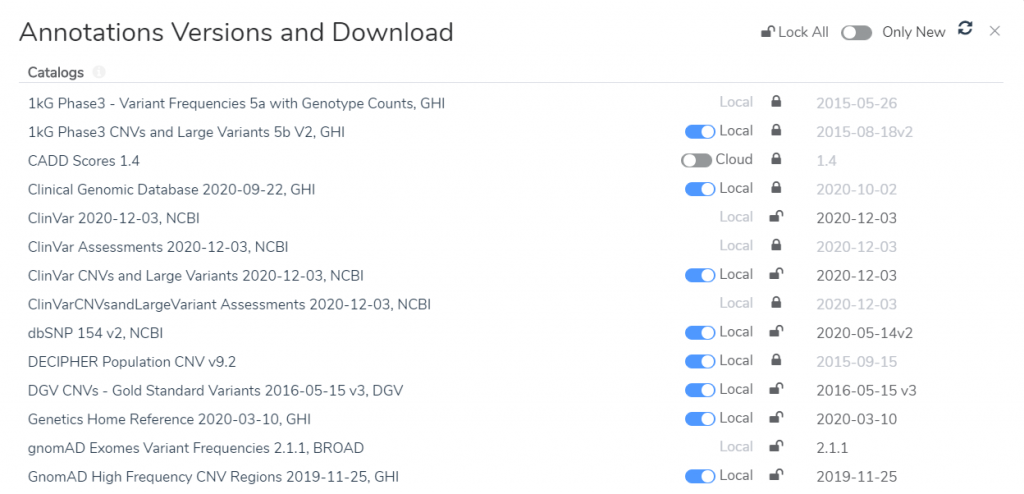
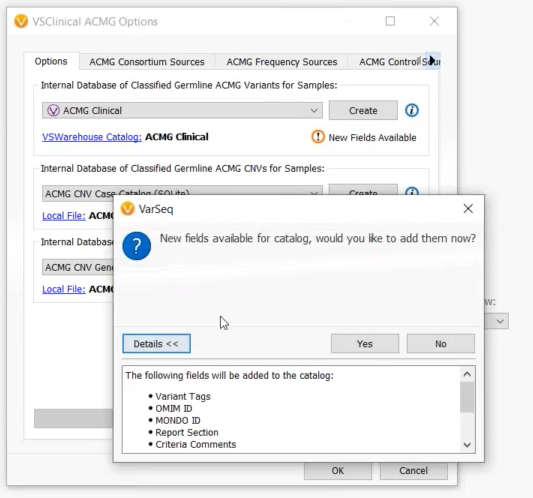
The VSClinical ACMG catalog schema has been expanded with fields “Omim ID”, “Mondo ID”, “Report Section”, “Criteria Comments”, “Citations Data”, “Interpretation Citations”, “Exon Number” and “Interpretation Notes”. There is a useful button in the ACMG configuration dialog to upgrade your catalog to include these fields. Once upgraded, all newly saved interpretations will include data in these fields from the evaluation.
The updates to the ACMG Guidelines have now been incorporated into the ACMG workflow, many of which are changes to the recommended criteria and scoring strengths.
- The PM2 criteria recommendation is now based on a very low allele frequency instead of an absolute total allele count to be more robust to population catalogs with small sample sizes. In rare cases, the previous behavior resulted in both PM2 and BA1 being recommended. These settings are tunable, but by default the PM2 threshold is less than 0.02% for recessive genes and 0.01% for dominant genes. The threshold will be evaluated on the sub-population with the largest allele frequency that meets the specified minimum allele count.
- The ACMG guidelines have been updated to incorporate the Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. This allows PVS1 to be recommended at lower strength levels (Strong, Moderate, Supporting) based on LoF variants with less definite evidence of pathogenicity.
- The Gene Constraints track used by VSClinical has been switched from ExAC to gnomAD. You can expect the constraint values used in PVS1 and PP2 to evaluate the tolerance of loss-of-function variants and missense variants (respectively) to be different. Additionally, the LoF value now being used is based on the computations of the observed/expected ratio (O/E) and specifically the upper bound of the 90% CI computed on this value as suggested by the gnomAD team in the gnomAD v2.1 blog post.
- A special consideration warning is now given if “PVS1” and “PP3” are both scored.
- The ACMG criteria have had the evidence strength modifiers expanded to include newly published papers’ recommendations.
There have also been several enhancements to the ACMG Classifier algorithm.
- Now, adding the ACMG Classifier algorithm will always use the existing project’s version of the gene track for nearby variant criteria in order to stay consistent with the current project. Previously, the algorithm always selected the most recent locally downloaded gene track version.
- The ACMG classifier algorithm now ensures that previous classifications are detected and counted even when the transcript name has changed, such as when a gene track has been updated.
- The ACMG classifier algorithm now matches any gene annotation algorithm in the project to define the splice region variant for the purpose of the variant sequence ontology.
VSClinical AMP: Features and Polishes
The AMP workflow report templates have been updated to use the new customizable “filters” technique also used in the ACMG workflow. This means the input data has been re-organized to be less redundant while also providing more details.
A “Somatic Catalog” was added to the AMP workflow. This allows saving and auto-filling previously saved oncogenic scoring and interpretations at the variant level for somatic variants. You can save/load variants into this catalog in the “Variants” tab, while still using the “Cancer Interpretations” catalog in the “Biomarkers” tab.

The cancer tumor type options have been expanded to have a top-level type for each tissue type. For example, “Lung Carcinoma” can now be selected instead of just one of its sub-types.
When searching for clinical trials, there is now a filter option for trials that contain specific Diseases in their summary.
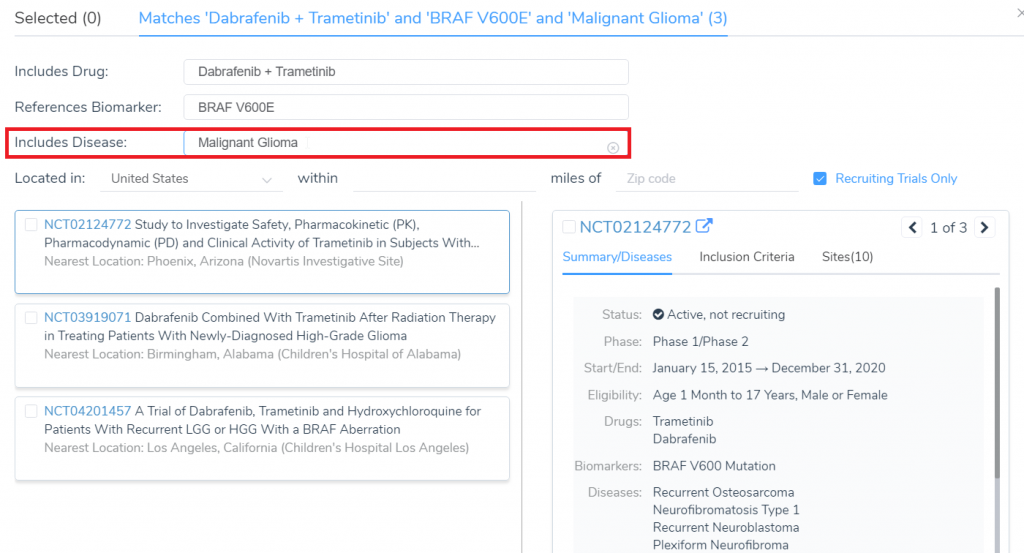
This was one of the larger VarSeq releases containing many new features and bug fixes. A full list of all the changes in VarSeq 2.2.2 release can be found here. We have been generating and will continue to create blogs that go into detail describing how these features can integrate into your workflows so stay tuned in the blog! Please reach out to us at support.goldenhelix.com if you have any questions or run into any issues as you integrate these new features into your workflows.