VarSeq 2.2.1 was released on April 1st and features an upgraded gene annotation capability with new RefSeq genes tracks and an AMP workflow addition: the Drugs and Trials tab.
The new RefSeq human genome genes tracks contain updated gene names and the recognition of any MANE (Matched Annotation from NCBI and EMBL-EBI) identified transcripts. VarSeq has been updated to be able to recognize the new names as well as the old names (or Aliases).
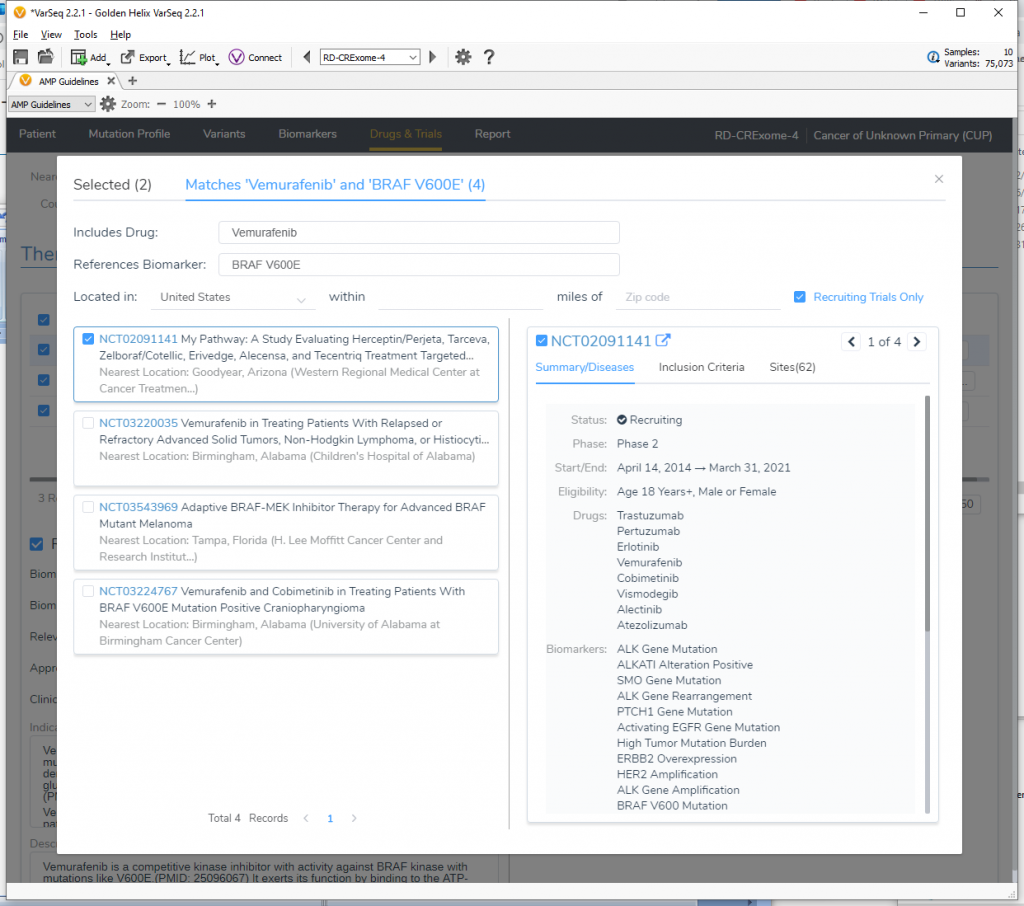
The new Drugs and Trials feature allows users to locate different clinical trials all over the world and even lets them search within relative distances from certain zip codes in the US. Check out our recent webcast “A User’s Perspective: Drugs & Trials for Cancer Diagnostics” highlighting this capability.
Another new noticeable feature is the ability to lock or unlock annotation sources in templates. For example, if an annotation source like ClinVar was locked (the default option) the version of ClinVar will be used when the template is used-hence the version is ‘locked’ in that template. If that annotation source is unlocked and a new version of ClinVar is available when the template is used next, that most recent version of ClinVar will be used. This general trend of updating annotation sources was also carried to VSPipeline. Now there is a command to update annotation sources when using VSPipeline.
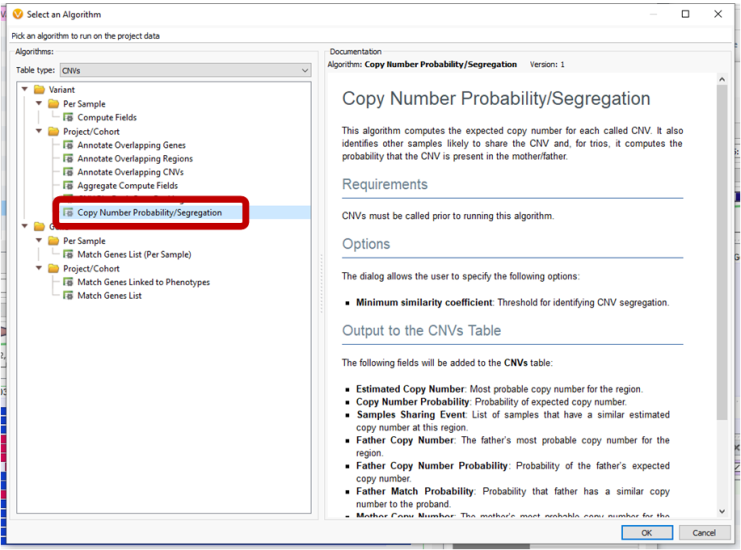
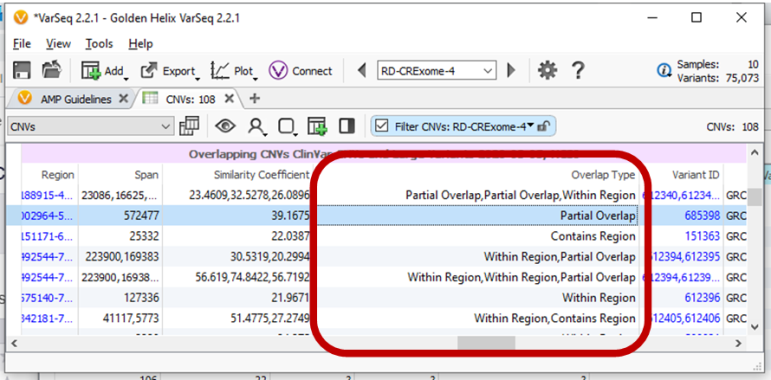
Other new features in the CNV realm include a CNV Probability/Segregation algorithm which looks at the probability of a CNV existing in a parent sample (like with trio analyses), and an Overlaps field so that when CNVs are annotating, the output will tell if the CNV is fully contained or partially overlapped.
New Features
The new VSClinical AMP workflow has been updated with a new Drugs & Trials section to support the inclusion of details on relevant therapies and clinical trials in the produced report.
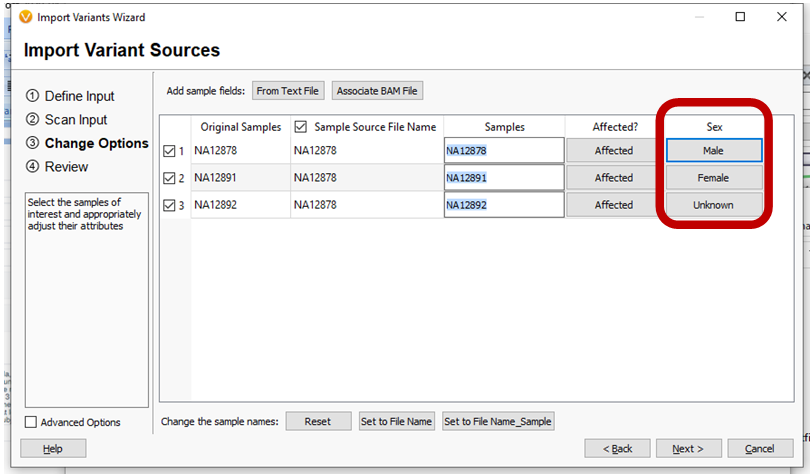
A new field has been added to the Import Wizard so the Sample’s Sex can be specified on import.
There is now a remote staging server that contains annotation sources that are in development but are not yet ready for regular release. You can add it in the Manage Data Sources dialog by selecting Remote and Staging Annotations from the New locations dialog.
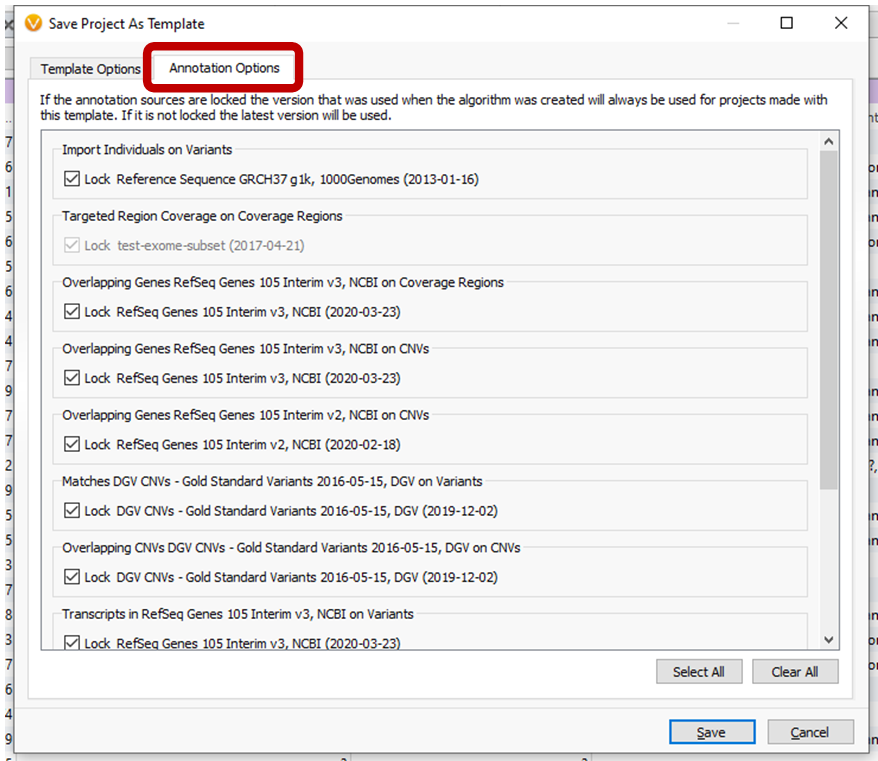
Saving projects as templates from active projects now gives users the option to automatically update annotation sources on new project creation for select sources, or to preserve the annotation versions. See the Annotation Options tab of the Save Project as Template dialog.
There is now a new command option in vspipeline to update annotation sources in the project template to the most current annotation source before importing data.
A new CNV Probability/Segregation algorithm was added for trio models to show the expected copy number for each called CNV as well as the probability that the CNV is present in the father or mother samples.
Annotating CNVs with CNV catalogs or other interval types now includes a new Overlaps Type field. This enables filtering of whether the current project region or CNV is fully contained within the annotation or fully contains the annotation.
For CNVs annotated against genes, the HGVS and Sequence Ontology are provided as additional fields for CNVs that are contained within a gene. This provides useful details about whether the deleted or duplicated exons cause frameshift or in-frame changes to the protein sequence.

Bugs Fixed
- On VCF import, the default behavior to “fill-in” fields at the same genomic location across normalized records in specific situations overwrote values that were present from other VCF lines for the same samples. The information from the VCF is now consistently conserved.
- Samples that were manually renamed within an active project were keeping their original names in the VSWarehouse upload. This has been changed to preserve the changed name.
- Subsetting down samples to be uploaded to VSWarehouse was causing a crash. This has been changed to allow sample subsets to be uploaded.
- When lifting over data on import or in data curation, some data was not correctly adjusting for when the reference and alternate allele switched. This has been fixed.
- There were issues accessing MySQL databases for storing assessment catalogs on Mac platforms. The specific database driver was updated to fix this issue.
- In CNV tables, the Region column was not able to be changed to different display modes such as Chr, Start and Stop without being hidden. This has been fixed.
- Setting AppData folder file paths to top level network drives was causing an issue, but this is now supported.
- The Kinship fields produced from running the Sample Statistics algorithm on a family trio were not respecting the family relationship if the proband was not the first sample. This has been fixed, and also parental relationships can be changed, and the Sample Statistics will rerun in an open project to reflect any changes.
- VarSeq would sometimes fail to load the annotation repositories when connected to a warehouse annotation server that caused a timeout. This issue was resolved, and the network timeout was raised to two minutes for loading remote annotation sources.
- The Sample PhoRank Gene Ranking algorithm was incorrectly linking phenotypes to subsequent samples. This has been changed to not propagate previous gene sample scores.
- The following changes have been made for VSClinical AMP Guidelines:
- In the Biomarker Summary Assessments tab, CIViC records under the same CIViC Variant ID will be collapsed into one assessment entry with the highest star rating assessment displayed.
- After revoking a Report sign out in the AMP Guidelines, the Report field for Small Variants on the Mutation Profile tab could not be adjusted. This has been changed so Small Variants can have the Report field changed after revoking a report.
- The Allele Ratio bar section in the sidebar of somatic variants was not showing the ratio amounts for project variants.
- The following changes have been made for VSClinical ACMG Guidelines:
- The genotypes of the parent samples in a trio project were not being read correctly when added to VSClinical. Now the genotypes and inheritance pattern is filled in from the project.
- Leaving VSClinical and the Available Downloads dialog open for an extended period could use more computer memory while searching for download dependencies. This has been fixed to not grow memory usage over time.
- When a variant is not present in gnomAD, a link to open the region in the gnomAD coverage browser was not working with the latest gnomAD website. The hyperlink has been updated to generate the right types of intervals.
Polishes
- Altering the location of an AppData Path or Folder in VarSeq used to require a refresh in the Manage Data Sources… window to locate the new file path, but now this is updated automatically when a new instance of VarSeq is opened.
- The Match Gene List function now includes an option to load gene name values to auto complete list entries and to confirm that the gene name is recognized. This helps ensures that all entered genes are valid gene symbols. This feature leverages the new Aliases field present in the updated RefSeq genes tracks.
- After running, the Match Genes List function will list gene names that were not found in the gene source list in the run log and the source information details. Additionally, the gene or alias association is displayed in the curation notes section.
- The HGVS notation for a variant for insertions edge-case has been improved. Duplicate codon insertions are now described in their short form (such as T599dup versus T599_T599dup) and insertions that include a stop codon now match the updated HGVS specification to describe in the inserted sequence (i.e. p.E858_L859insVP* instead of p.L859_W1755delinsVP).
- The dialog listing out of date sources that may be updated now additionally lists which table those sources are on when they are not on the variant table.
- The RefSeq Genes annotation tracks now include a MANE Status field to indicate if the gene has a MANE identified transcript. If so, that transcript will be selected as the clinically relevant transcript.
- Importing data into an assessment catalog from a file, the Last Edit field had very rigid data format requirements. This has been adjusted to support different date formats.
- Improvements were made to an edge case when logging and the software remembers being activated for a different machine. This case is handled without requiring explicitly logging out or going outside the software to delete the user properties file.
- For the annotation Convert Wizard, liftover has been improved to better handle variant deletions and a new option was added to append the reference allele if missing on liftover.
- There is now an extra option to export the exon segments of downloaded gene tracks in a BED file format. This is useful for creating target panel lists for specific genes that reflect the coding regions of genes.
- Now the Log tab will denote the name of the template used in the project creation.
- The Convert Wizard options are now displayed in a tab style to condense the view.
- The browse for BAM path dialog has been updated to utilize the native Windows explorer dialog.
- Now the “Launch Another VarSeq Instance” option is available on all platforms and can be found under the File menu. (Previously only on Mac)
- The default VSWarehouse URL can be configured using a “hosts.json” file for deploying VarSeq to a large number of users.
- VarSeq now supports VCF.BGZ file extensions in the import process as an expected extension.
- VarSeq now allows users to specify a maximum number of processors to be used during computation. This feature can be found under Tools > Memory and CPU Settings.
- Now unicode characters can be used in sample names and in assessment catalog that save records per Sample, such as the ACMG classification catalog.
- The following improvements were made for the CNV algorithm and its outputs:
- The CNV Reference Sample selection algorithm has been adjusted to favor the most recently added samples with the most similar mean sampling depth. This prevents over-fitting when many reference sequence samples are available.
- When the CNV Caller was run using samples with an undefined percent difference or a missing BAM file path it would error out. Now it completes and skips the absent samples by filling in the corresponding output with missing values. A flag of “No coverage information” is placed in the Samples table for any skipped samples.
- Now the Annotate Overlapping Genes algorithm for CNVs will list all of the overlapped transcripts where before only partially overlapping transcripts were listed.
- The LoH Caller algorithm now provides the ability to change the Minimum Variant GQ value for calling LoH regions. Also, the default value for the minimum variant GQ value is set to 30 (it was previously 80).
- The icon to Add CNV Annotations was missing from tables of imported CNV and has now been added to match other tables.
- The following polishes have been made for VSClinical:
- A new field called “Other Transcript Effects” was added to the ACMG Guidelines table output that denotes effects present in neighboring transcripts for the same gene as the selected variant.
- Some language in our “recommendations” used one-letter HGVS and others three-letter HGVS for variants. This was changed to one-letter representation uniformly.
- The following polishes have been made for VSClinical AMP Guidelines:
- Reference genotype variants from callers like Freebayes can now be added as somatic variants to VSClinical projects.
- Now clinical trials are listed in the evidence tab for Biomarkers.
- Germline variants now have the Inheritance/Variant Type filled in for the Variants tab for germline mutations.
- Users will now be notified that a tumor type has not been selected when first selecting one of the add variants options in the Mutations Profile tab.
- Now the GenomeBrowse variant view will adjust to zoom and highlight the variant or biomarker selected.