Those of you who have been attending our recent webcasts have learned about our upcoming VarSeq release. A part of that release will be an additional algorithm that will annotate variants matching the current sample. If you are not familiar with these webcasts, here are several on-demand webcasts I recommend to get you familiar with these new features:
- Evaluating Copy Number Variants with VSClinical’s New ACMG Guideline Workflow
- A User’s Perspective: ACMG Guidelines for CNVs in VSClinical
The new tools will carry many users forward, by leaps and bounds, with the interpretation and classification of Next-Gen Sequencing based CNV results. By following intuitive guidelines, users work their way through variant analysis and match variants to the currently assessed sample. The VarSeq assessment catalog stores the processed variants, which include the single nucleotide variants, indel regions, and CNVs. Variants with prior evaluation, classification, and interpretation can be looked up and reviewed. Figure 1 shows the menu option for selecting the variant matching the current sample algorithm. This is would be based on the VCF level data (SNVs and Indels).
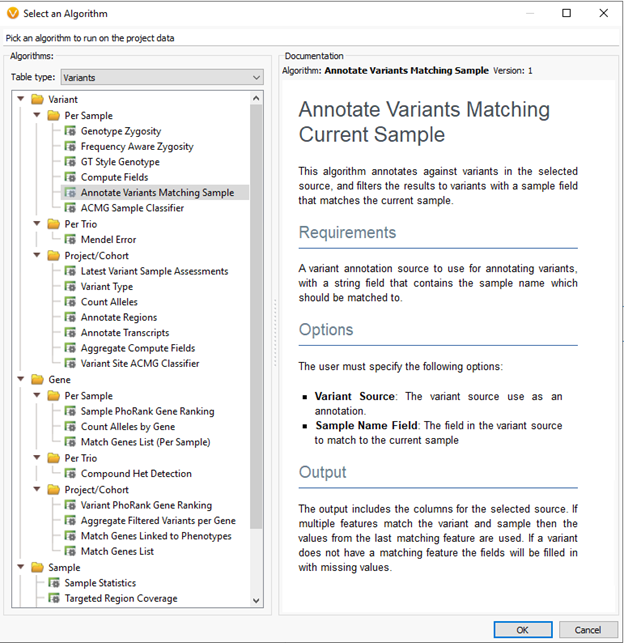
Generally, users will store any variants processed via VSClinical across all samples over time. By default, annotating against this assessment catalog will show the last variant submission for the last sample reviewed. For example, let’s assume over time you see the variant across many different samples. Initially, the variant was classified as uncertain significance. On the latest sample, the classification changed to pathogenic, based on some new evidence. If you want to go back and rerun the previous samples, you can use this tool to pull up the unique variant submission for that specific sample. From there, users can review the variant with the latest knowledge, update the classification, and record it for each sample. The variant level records, regions for biomarkers for somatic variants (Figure 2.), and copy number variants (Figure 3.) all utilize this tool.
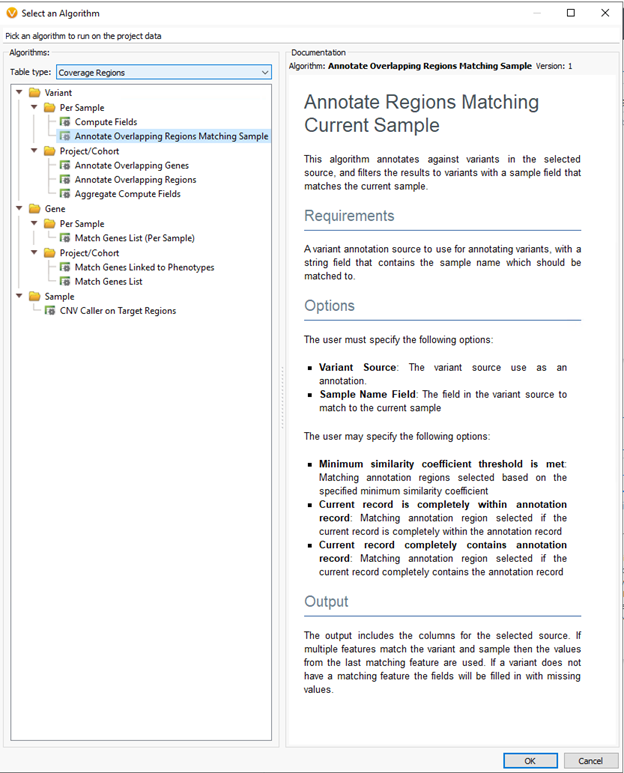
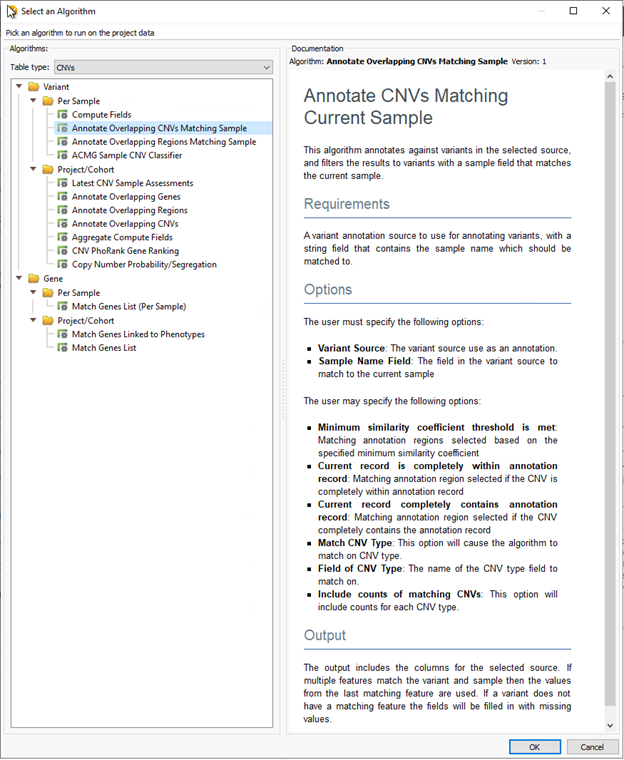
We are very excited about this upcoming release and look forward to sharing more details and resources with you. Subscribe to our blog to keep up to date on the latest news and updates coming from our team. Of course, if you would like to see these tools in action, please reach out to info@goldenhelix.com to schedule a demo or training with our latest beta builds.