I learned about Batten disease from a childhood friend’s Facebook post. Over the course of a few months, her 8-year-old, Eva, the oldest of 4 daughters – Emily, Lucy, and Carly – was rapidly going blind. Baffled, doctors ran a genetic panel that returned a devastating result – the diagnosis of Juvenile Neuronal Ceroid Lipofuscinosis or Batten disease. A broad class of rare fatal neurogenetic disorders that affect lysosome function, mutations of the CLN gene prevent the cell from proper waste disposal, causing a buildup of cellular garbage resulting in blindness, seizures, ataxia, and neurodegeneration. As the result of autosomal recessive mutations, symptom onset occurs between 5 and 8 years of age. As the disease progresses, dementia and paralysis require 24-hour care. Life expectancy ranges from early childhood to the mid-20s. For reasons still unknown, affected females experience more severe disease, losing independence and succumbing sooner than males (Cialone et al., 2012). Soon after her diagnosis, Eva started having seizures and motor impairment. Her parents waited in agony for Emily, Lucy, and Carly’s test results between Eva’s trips to the hospital.
In the case of an autosomal recessive disease like Batten, Eva, Emily, Lucy, and Carly each had a 1 and 4 chance of inheriting both defective CLN copies, one from each unaffected carrier parent (https://www.ninds.nih.gov/disorders/patient-caregiver-education/fact-sheets/batten-disease-fact-sheet). They also stood a 50% chance of becoming a carrier like their parents, possessing the potential to pass the CLN mutant on to their own children or in combination with a paternal mutant, the disease itself. Despite the 1 and 4 odds of Eva being the only affected, Emily was subsequently diagnosed with Batten’s, Lucy was found to be an unaffected carrier, and Carly had inherited 2 normal CLN copies.
In the future, Lucy may want to start a family, but her decision could be impacted by knowing she carries a mutant CLN copy and having witnessed Eva and Emily’s battle. Believed to affect 2 to 4 children out of every 100,000 in the U.S., Lucy may want to know if the same CLN mutant lurks in a paternal genomic contribution or include the CLN mutant in a perinatal panel. Pre- and perinatal panels are becoming more affordable and accessible as NGS continues to become cheaper and faster – in addition to more data-rich, generating more sequences with higher coverage. The challenge becomes sifting through a mountain of data and, if present, finding that clinically relevant variant of CLN.
The solution? A well-constructed filter logic that not only includes standard quality filters like read depth and coverage but filters for functional predictions, phenotypic associations, and classifications (such as ACMG or AMP). In this example project, we’ll look at a sample from a project of 54 exomes with 144,420 variants and go step-by-step to identify that key variant, a CLN mutant for someone like Lucy.

Step 1: Filter Variants Based on Quality
Before we can start finding those interesting and clinically relevant variants, we want to make sure that we’ve removed poor-quality variants. Here, I’ve set up a filter container called “Quality Filters” just for that purpose which includes Read Depth (DP) and Genotype Quality (GQ). Given that we’re working with exome data, we’ll set DP to 100, keeping only the variants whose read depth is equal to or greater than 100x. Next, we’ll set our GQ to a pretty stringent 50, selecting only those variants whose Phred score gave a 0.001% chance of an inaccurate base call. After filtering on DP and GQ, you’ll see that we’ve gone from 144,220 down to 18,089 variants based on quality alone.

Step 2: Identify Rare Variants
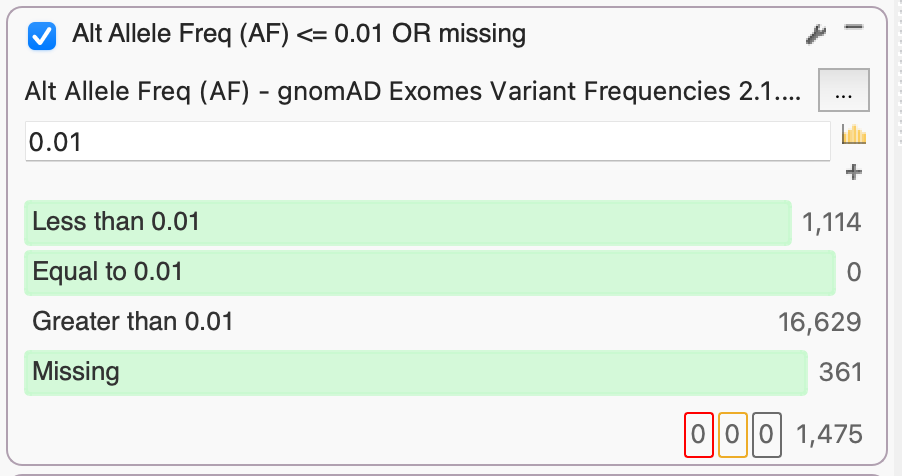
By leveraging several population catalogs available in VarSeq, such as the Genome Aggregation Database (gnomAD), we can use an aggregation of exome data to identify rare or missing alleles based on alternate allele frequency (AF). Here, we used gnomAD Exomes Variant Frequencies that currently include 125,748 exomes from unrelated individuals. By setting an AF threshold of 0.01, we select the 1,475 rare variants that occur in 1% or less of gnomAD exomes or are missing entirely.
Step 3: dbNSFP Functional Predictions
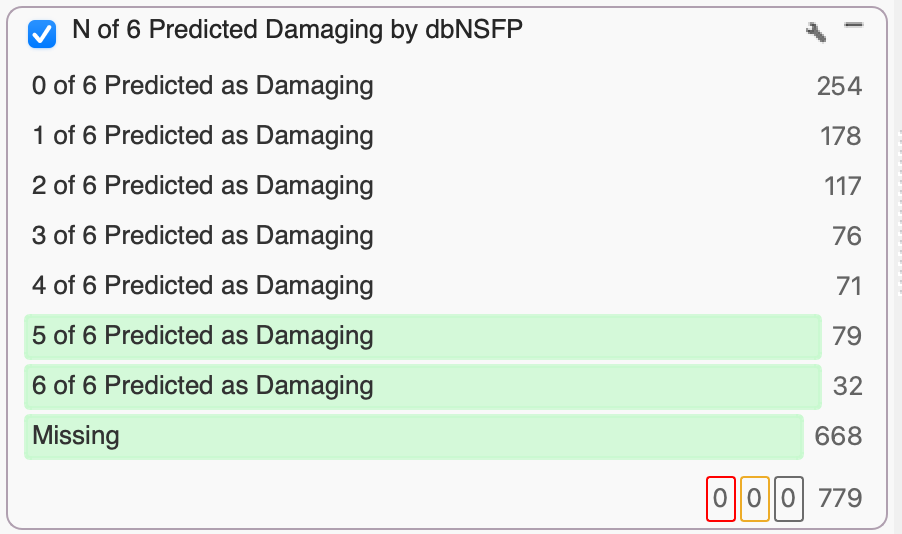
The database of Non-Synonymous Functional Predictions (dbNSFP) Voting provides, you guessed it, a vote. The vote is the number of independent functional prediction algorithms that anticipate a given variant to be tolerated or damaging, based on the impact of amino acid substitutions on protein function and the number of conserved nucleotide positions. These algorithms currently include SIFTPhred, Polyphen2HumVarPred, MutationTasterPred, MutationAssessorPred, FATHMM Pred, and FATHMM MKL Coding Pred.
If you want a conservative filter, simply keep things with 0, 1, or maybe 2 Tolerated predictions. A more conservative filter would keep variants with 3, 4, or 5 Damaging predictions. In this example, we were extra conservative and selected variants with 5 and 6 Damaging predictions. While many variants won’t have 5 algorithms with non-missing values, a soon-to-be-released dbNSFP will expand the vote with new functional prediction algorithms, so stay tuned!
Step 4: Use PhoRank to Rank Genes Based on Phenotype
Now that we’ve narrowed in on 779 variants, we can utilize the Phorank algorithm to identify and rank genes relevant to a user-specified HPO phenotype, which can be extended to include phenotypes and syndromes from OMIM. Developed by our very own Golden Helix engineers, PhoRank takes the best of the Phevor algorithm, which assigns scores to ontology terms, or seed nodes, based on their proximity to that user-specified phenotype, and weighting these nodes based on similarity – i.e., discrete nodes that are highly related to the specified phenotype get a higher score. In comparison, more general nodes with a lot of neighbors get a lower score.
In our case, we’re interested in genes associated with Batten disease, also known as neuronal ceroid lipofuscinosis. Batten has 13 subtypes, so we could select specific subtypes, but any combination of mutant copies will result in disease, so we’ll select all 13. Once the algorithm runs, we can set a gene rank filter threshold. Given Lucy’s family history and affected status, we set a high threshold of 0.95, selecting variants with a 95% or higher association with Batten. Depending on the user case, a lower threshold might be more appropriate and equally informative.

Step 5: ACMG Variant Classification
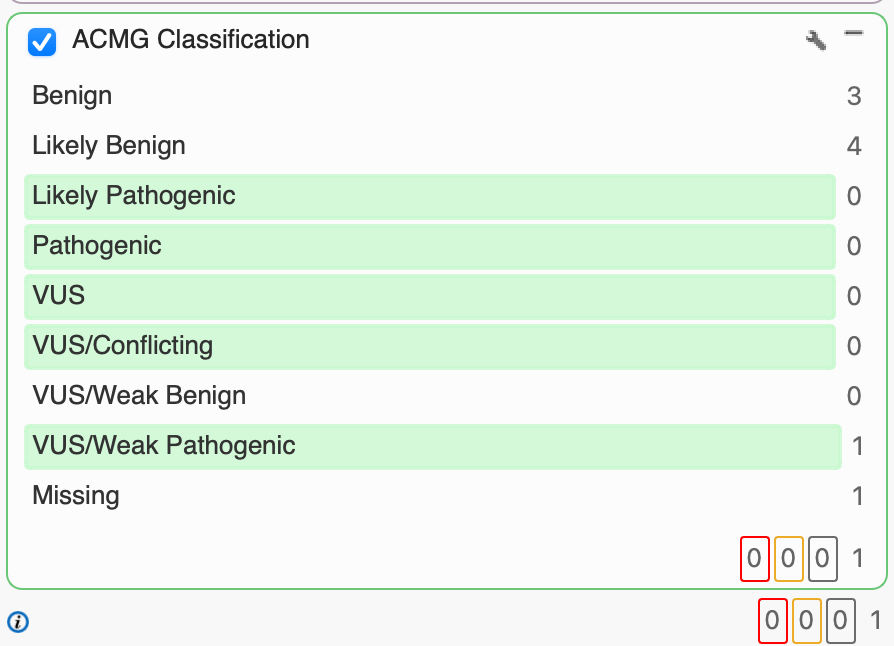
Having used PhoRank to identify 8 variants with a 95% or greater association to the 13 Batten subtypes, we can leverage our ACMG Classification algorithm to classify each according to the 33 criteria that make up the ACMG guidelines. Classification is based on evidence, including population frequencies, conservation scores, splice site algorithms, and functional predictions. For our purposes, we’re interested in variants that have been classified as VUS, VUS/Conflicting, VUS/Weak Pathogenic, Likely Pathogenic, and Pathogenic. Having applied the ACMG Classification filter, we’re able to narrow in on one particular variant of CLN8 with a VUS/Weak Pathogenic classification rather than investigating the other 7 Likely Benign or Benign variants.
Having identified a clinically relevant variant for Batten disease, check out our other blogs and tutorials to learn how to assess a variant like CLN8 in VSClinical. You can explore the detailed rule logic rule and supporting literature used to classify a variant according to ACMG guidelines, how to incorporate this information into your interpretation with the click of a button, and incorporate it all into a final clinical report – equipping someone like Lucy and her doctors with life-changing information.