In my recent webcast, I demonstrated how VS-CNV users can detect high-quality copy number variant events. If you didn’t have a chance to join, you can view the recording below!
This webcast generated a lot of great questions! If you have any other questions about the content covered in this webcast that
Do you have a sense on how the actual sensitivity and specificity performance of the algorithm is at <0.001?
If there is any concern that you are losing possible true events with the precision setting, you can always run the sensitivity setting or balanced to capture more by increasing the probabilities for copy number variant detection.
Do you have recommended criteria for filtering on z-score and ratio?
There are specific ratio and z-score thresholds already in place for calling the copy number variant event. You wouldn’t necessarily need to manually filter on these metrics as you could potentially eliminate true copy number variant events.
That said, the Robarts paper, “Use of NGS sequencing to detect LDLR gene copy number variant in familial hypercholesterolemia,” used custom thresholds for ratio and
Do you take into account split reads in VS-CNV?
Currently, we don’t take into account split reads. Our method is designed for more straightforward coverage designed for panels.
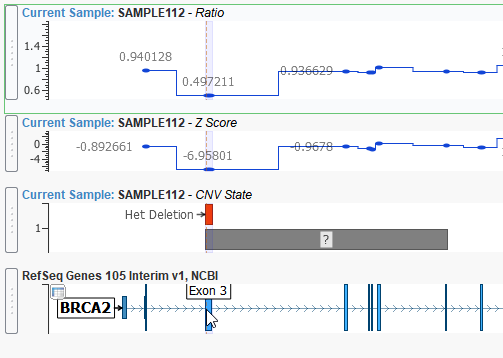
Can you speak to VS-CNV’s ability to call single exon heterozygous copy number gains and losses?
Since calling copy number variant events in VS-CNV is based on coverage data, our algorithm can call both large and small events including heterozygous copy number gains and deletion.
In the example below, we are displaying a heterozygous deletion event that covers exon 3 of the BRCA2 gene. This event spans 288 base pairs and is validated using our ratio and z-score metrics.
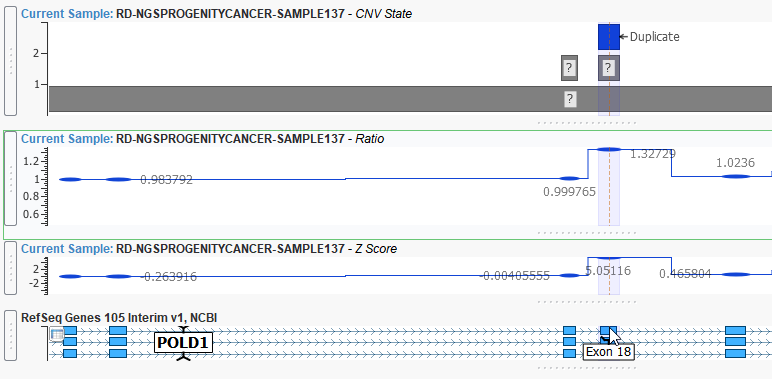
This event is an example of a CNV heterozygous duplication that spans 177 base pairs of exon 18 of the POLD1 gene. This duplication is reinforced with a ratio of around 1.5 and a
Further supporting VS-CNV’s ability to detect single exon events is the Robarts paper “Use of NGS sequencing to detect LDLR gene CNV in familial hypercholesterolemia.” This paper demonstrated 100% concordance between MLPA and VS-CNV’s ability to call a duplication event in exon 7 of the LDLR gene.
Assuming that I have called my CNVS and I have a .xslx file (or a cnv.vcf) file. Could I upload the file in VarSeq and annotate the copy number variants to generate a clinical report?
Unfortunately, you cannot import your copy number variant results into VarSeq directly as sample data. The computation of copy number variants in VarSeq is based on the detection of the events leveraging coverage data stored in your BAM file. However, you can use known copy number variant events as an annotation source which can be leveraged against the computed copy number variants in VarSeq and included in a clinical report.
How do you measure the percent difference (>20%) of references samples from tumor sample?
The percent difference of an individual sample to another is a reflection of the average percent difference of each individual target across all targets in each sample. In other words, the individual differences in your reference sample column and the percent difference is the average across all reference samples. These two columns can be found in the samples table under the Copy Number Variants fields.
I am now a current user of VS 2.0.1 and am working on the exome trio template. However, while filtering through variants, I discovered that somehow the GQ field in my data was missing. I would like to inquire why is this, and also how will this affect my results?
Likely this is a case where a variant exists in a different sample. The first thing to verify when looking at a sample field with missing data is if that variant exists in a different sample. One way to do this is by field collating all of the samples to verify if the variant exists in one sample but not the other.
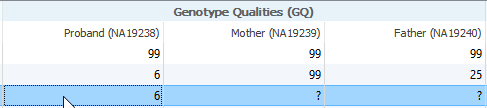
What about the pseudogenes?
The fundamental basis of detecting copy number variants with VS-CNV is from the coverage coming from the BAM file. Regarding pseudogenes, it will add another layer of complexity to what you are aligning to. That said, please feel free to contact us if you would like to evaluate some of your data.
What do all the grey bars with question marks refer to?
The grey bars with question marks refer to copy number variants in other samples. To identify copy number variants specific to a sample, you will need to utilize the filter chain to filter down true copy number variant events present only in that sample. This primarily includes adding the copy number variant state to the filter chain and selecting Deletion, Duplications, and Het Deletions.
Generally, is VarSeq suitable for analysis of germline cancer samples?
Absolutely! Here is a link to our recent publication https://link.springer.com/protocol/10.1007%2F978-1-4939-8666-8_9
Is there a maximum number of samples that one can evaluate at a time?
There is no maximum threshold. However, you may consider managing your reference set to ensure you are detecting high-quality CNVs. Fundamentally, you could build a project with your references imported, to asses if there are any low-quality samples needing to be removed from the reference set.
Can I associate BAM files from the cloud?
Unfortunately, no. The BAM files will need to be stored in a location that the software can read/write to the associated folder/directories.
How can I compare copy number variants events identified across samples with VS-CNV?
You can plot metrics such as the average ratio and z-score for all samples and change your display option to view all copy number variants shared across all samples.
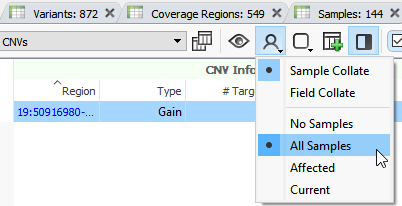
Can VSClinical annotate CNV.vcf files to ClinVar and generate reports?
We don’t currently have the capabilities to evaluate copy number variants with VSClinical. Also, you will not be able to import CNV.vcf files as sample data in VarSeq, but you can leverage the information as an annotation against detected CNVs computed in VarSeq. Also, you can include CNV results in the clinical reports in addition to variants evaluated in VSClinical.