Thank you to those who attended our webcast on the user perspective of our automated AMP guidelines! Furthermore, let me express our appreciation for those particularly engaged users who posed some very thoughtful questions. While we weren’t able to answer all of them live, I hope to shed some light on some pertinent details of somatic analysis here.
Let’s start out by answering some workflow questions, the first of which pertains to how to leverage variant allele frequency/fraction (VAF) in our filter chain. For those wondering why we chose a cutoff of 0.01 in our demonstration, there are a couple of things to consider. First and foremost, we should reiterate that this choice is completely modifiable depending on the requirements of a given workflow. One good tool for determining what a good cutoff might be is the histogram tool present on the filter card (Figure 1):
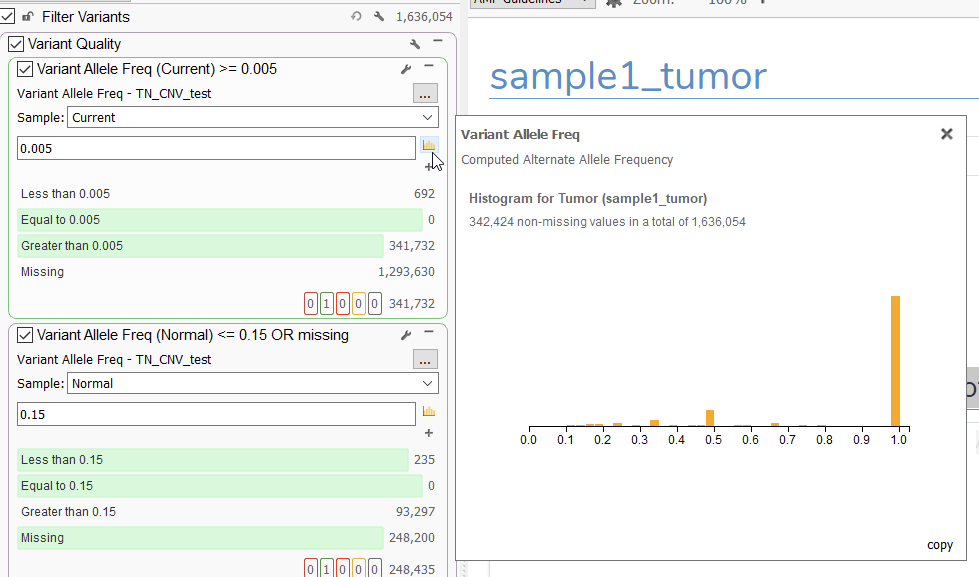
Here, users can visualize the spread of VAF values in a sample, which can be used to determine a good cutoff for the workflow.
Filtering on VAF is also a consideration when it comes to ruling out probable germline variants. While we can encounter somatic variants with a continuous VAF from 0 to 1, we would expect to see germline variants at ratios of around 0.5 or 1.0, barring CNV considerations. 0.5 would be more indicative of a heterozygous state, and 1.0 would be indicative of a homozygous state for the variant. Hence, by providing a low cutoff for somatic VAF, we set ourselves up to analyze a larger proportion of probable somatic variants. This publication from BioMed Central provides some useful insights and caveats pertaining to VAF.
This leads to the question of differentiating between germline and somatic variants in VarSeq. The easiest way to do this, as we briefly touched on during the webcast, is with a tumor-normal pair workflow. This strategy allows us to rule out variants present in our normal sample as germline and filter down to variants only present in the tumor sample. Beyond this, there are several other filters we can apply that increase the likelihood of removing germline variants, including our inverted ACMG auto classifier filter, which eliminates obvious benign variants. All of that being said, there is, of course, no way to know with 100% probability that a variant is somatic via filtering, so depending on how holistic you want your search to be, we recommend fine-tuning the various thresholds and filters applied as you validate your workflow.
Find out if VSClinical is right for you.
Another excellent question we had related to the filtering strategy is that of finding rare or novel somatic variants that are not well-documented in CancerKB or other databases. The main caveat I’d like to address here is the philosophy behind the AMP guidelines. That is, they are intrinsically used to classify well-documented variants, so fundamentally, we’re considering a different application when it comes to rare variants. There are nonetheless a couple of considerations for this type of workflow.
One option is to include a parallel filter chain that is more holistic. Filters pertaining to variant quality, population frequency, and sequence ontology will still lend themselves well to the search for novel somatic variants. However, you’d probably want to invert the logic we explored with filters like COSMIC and instead look for novel variants. Importantly, these are some very brief considerations; for a more in-depth look at specific use cases, the best resource is our FAS team, myself included. We are more than happy to schedule a training session to explore specific workflows tailored to rare somatic variants.
Another tangential solution is incorporating VSWarehouse into your software suite; as we discussed during the webcast, it is a great auditing tool for circling back to Tier III variants as more information becomes available.
We also had plenty of great questions about current and future VarSeq capabilities and how VSPipeline fits into the picture. As a general overview, just about anything that we don’t natively support can be brought into VarSeq and rendered into a final report either manually or automatically with VSPipeline. Any information included in a text manifest used in tandem with VCF importation is automatically included in the JSON output of VSClinical, and can then be pulled into a customized final report (Figure 2). The addition of a text manifest can be done automatically through VSPipeline or manually during the importation step. For more details on report customization, check out our webcast on advanced report customization, or schedule a training session with us!

I’d also like to briefly touch on some more specific items of interest. Unfortunately, VarSeq does not support any kind of RNA variant analysis. However, we currently have the ability to manually evaluate fusions and can include considerations like microsatellite instability and tumor burden via a text manifest, as discussed in the previous paragraph. Furthermore, users can look forward to direct TSO 500 implementation in a future release of VSClinical, for which we are very excited.
Again, I’d like to thank all of our attendees and customers for directing our development process. If your question wasn’t directly addressed here, or if you have any additional follow-ups, please don’t hesitate to reach out to us at support@goldenhelix.com. I look forward to furthering our correspondence.
VSClinical allows for the clinical interpretation of variants based on ACMG & AMP Guidelines. As an optional add-on, VSClinical can follow the Association for Molecular Pathology (AMP) guidelines used for performing targeted cancer gene panel tests popular in molecular pathology labs to report significant biomarkers of clinical utility in precision medicine. The AMP guidelines for the interpretation of somatic variants provide a set of best practices for evaluating and reporting variants for cancer genomic testing. VSClinical provides a guided workflow for following these best practices, including evaluating the clinical evidence for a reportable biomarker using the AMP Tier evidence levels. VSClinical provides a tailored workflow for interpreting and reporting small variants as well as CNVs and fusions. Contact our sales team for more information.
