Our upcoming release of VarSeq is one of the largest we’ve ever had with our software! It comes with an extensive list of polishes and new features like our recently mentioned ACMG CNV classifier and a redesigned reporting interface with updated templates. Additionally, this new release is also paired with some major upgrades to our list of new and supported annotations. One crucial track in any workflow is annotating variants against gene information in RefSeq.
The RefSeq update will not only factor for any new transcripts but will also include the following improvements:
- Gene names: Updated to match the latest in Entrez Gene
- Aliases: Added based on the latest information in Entrez Gene
- Summary of Product: Updated to match the latest in Entrez Gene
- LRG ID: Added to indicate which transcripts are in the Locus Reference Genomic database
- MANE Status: Added to indicate which transcripts are part of MANE Select 0.91 Release
Keep in mind this updated gene track is supported for both the GRCh38 and GRCh37 assemblies. Not only is this data posted in the variant table but many of the fields are working behind the scenes with our gene annotation algorithm and presented in VSClinical (Figure 1). One key component of this algorithm is the selection of the clinically relevant transcript. Many components impact this transcript selection such as the size of the transcript, how many clinical submissions are present, and lastly, the huge effort associated with the MANE select transcript based on matching RefSeq and Ensembl selected transcripts.
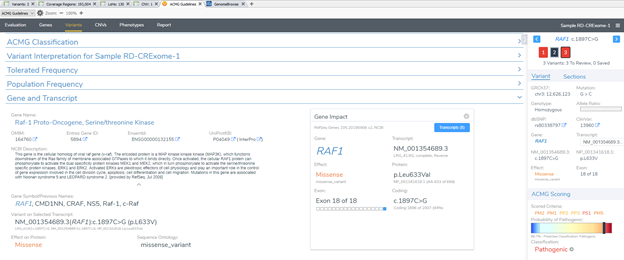
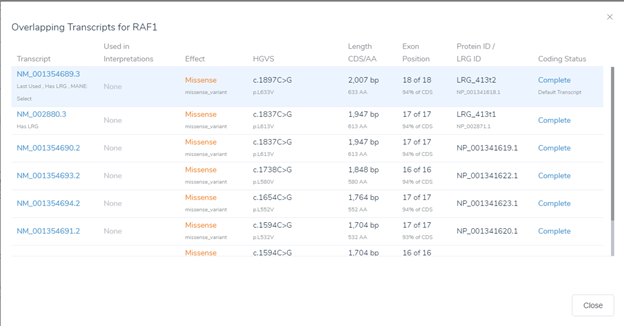
In any case, the user can still select their preferred clinically relevant transcripts and maintain the selection in a standard workflow saved in their project template. RefSeq plays a huge role in variant filtering and analysis for both the single nucleotide, indel, and copy number analysis, all processed in the VarSeq software. If you wish to explore more of this annotation or any others and learn to incorporate them into your workflow, please contact us at info@goldenhelix.com to schedule a training session. Here also is a breakdown of our recent release notes on version 2.2.2 Feel free to check out some of our other blogs that always contain important news and updates for the next-gen sequencing community.