With the release of VarSeq 1.3.1 we have included a new demo project to showcase a single tumor-normal pair analysis workflow. The project can be accessed through VarSeq and VarSeq Viewer by going to File > Open Example Projects > Example Tumor-Normal Pair Analysis.
This project contains an exome pair (Normal-N990005 and Tumor-T990005) from the Gastric Cancer study Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes published in Nature Genetics. Exome sequence data was downloaded from the NCBI Sequence Read Archive (SRA) under ascension number SRA045832. Batch variant calling for single samples was done using BWA + GATK through the Seven Bridges Genomics, Inc. pipeline. The full BAM and VCF file data is available for download through VarSeq by going to Tools > Manage Data Sources and selecting them from the Example Samples > Gastric Cancer Samples location.
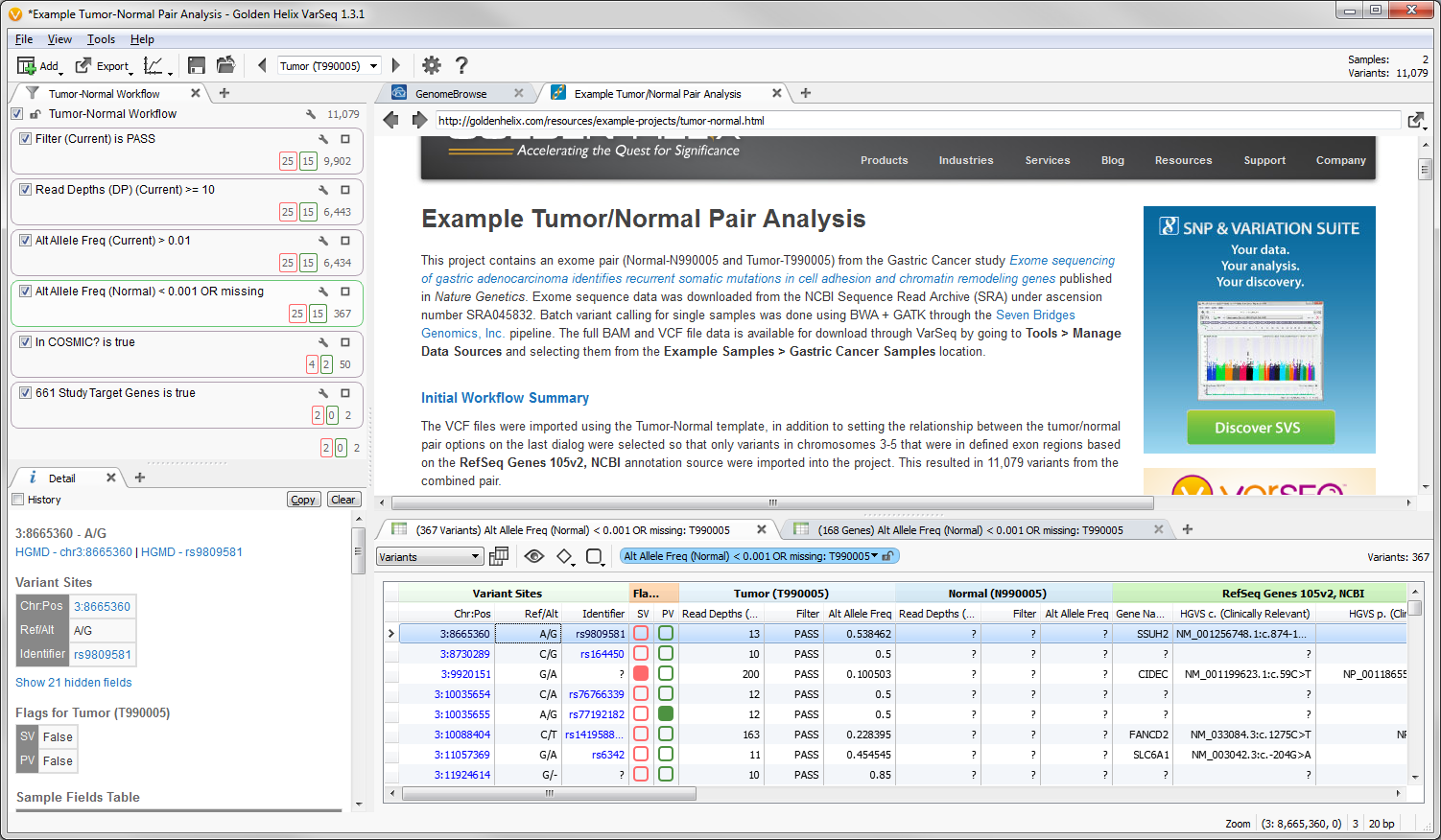
Fig. 1. Project view for Example Tumor-Normal Pair Analysis
The VCF files for this dataset contained over 2 million variants, so when importing the data into the example project we selected to only include those variants in chromosome 3-5 that were in defined exon regions based on the RefSeq Genes 105v2, NCBI gene annotation source. This resulted in a total of 11,079 variant for the combined pair.
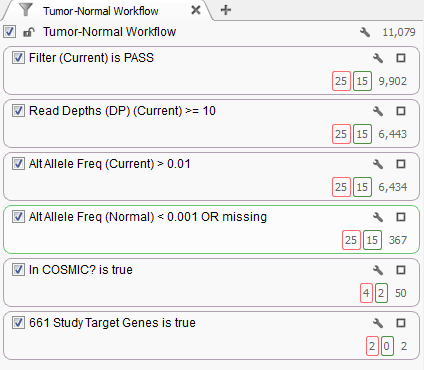
Fig. 2. Filter chain for the project
The filter chain for this project is based off the shipped Tumor-Normal template that is designed to identify variants found in the tumor sample at an allelic frequency greater than 1% and are only present in the normal sample at an allelic frequency less than 0.1%. Additional quality assurance filters guarantee the variants pass the QC measures from the variant caller and have sufficient read depth. After that, we will only consider those variants that have been annotated in the COSMIC database.
The last filter card in the filter chain was created by the Add > Computed Data… > Match Gene List algorithm which determines matches between the gene annotation of each variant and a user selected list of gene or identifier symbols. The original study for this dataset identified 661 genes that contained non-silent somatic point mutations and this list was used to create the filter criteria. Of the 50 variants that passed the filter criteria to this point 2 of them were found within these 661 genes.
Two Variant Sets were created to flag variants within the project, one for confirmed somatic mutations and second for potential somatic mutations. As you scroll through the list of variants in the table you can verify those that are true somatic mutations from those false positives by examining the reads in the BAM file for each sample in a GenomeBrowse view.
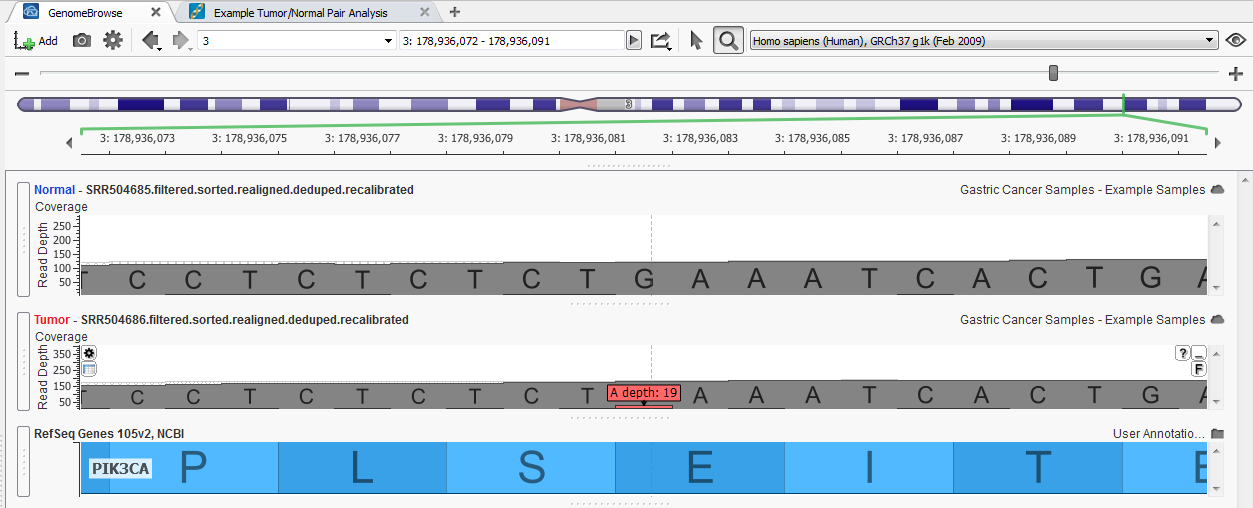
Fig. 3. Identified somatic mutation (3:9920151-G/A) within the PIK3CA gene
If you have any questions about these example projects or want to get a copy of VarSeq or VarSeq Viewer, please contact support@goldenhelix.com.