Our previous webcast from VP Gabe Rudy in September exposed us to some fundamentals of this years’ updated Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). This recent webcast was dedicated to breaking down these new guidelines… Read more »
Did you know you can control your preferred transcript settings for clinical interpretation in VSClinical? Your lab is analyzing the DNA of a tissue sample from a patient with small cell lung cancer. The lab technician has imported the patient data into VSClinical to detect clinically relevant variants and evaluate and score these according to the AMP Guidelines, as well… Read more »
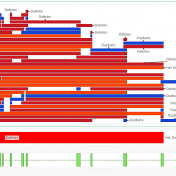
In this blog update, I’ll be walking you through some of the advanced plotting capabilities with GenomeBrowse. The strategy with any next-gen sequencing analysis is to filter down to interesting variants for either research or clinical conclusion. Golden Helix produces powerful software specifically tailored for this efficient and comprehensive search for interesting and clinically relevant variants. One additional advantage of… Read more »
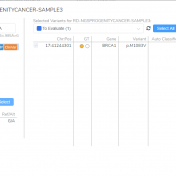
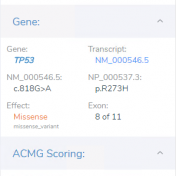
VSClinical is a feature to evaluate clinically relevant variants according to the ACMG or AMP guidelines. This feature can also be used to identify if a variant has been observed previously or evaluate a manually inserted variant. Take, for example, the scenario where a colleague is interested to see if you have seen any variants associated with Bechet syndrome, which… Read more »
In our recent webcast announcing the upcoming release of VarSeq VSClinical and the implementation of the ACMG guidelines for NGS CNVs, we had a number of live questions we didn’t get a chance to cover at the end of the presentation. I will follow up on those questions in this blog post. But first, if you didn’t get a chance to join us for… Read more »
In the 1990s the genetic industry voiced a request for a variant catalog that incorporates associated variant information such as phenotypic and metabolic pathways. The call was answered by NCBI, which created dbSNP; dbSNP became publicly available in 1998 and around 1.5 million variants. Fast forward to the present and dbSNP now contains over 2 billion SNPs spanning human, rat,… Read more »
We have now reached the final blog of the NGS-Solutions for Clinical Variant Analysis series. Part I of this series explored the capture of variant classifications in the VSClinical environment when following the ACMG and AMP guidelines. Part II was similar in content but for the capture of clinically relevant copy number variants as well as using a CNV catalog… Read more »
The potential of genetic testing to impact a patient’s life has been greatly accelerated by the sharing of variant interpretations done by clinical labs in public repositories such as ClinVar. This is not an inevitable outcome, but the persistent work and advocacy of people like Dr. Heidi Rehm and organizations like ClinGen. We recently participated in a survey and vetting… Read more »
SVS 8.9.0 was released on August 19th and features a new GBLUP by Bin feature and a new utility to find the LD scores of markers and categorize them into bins, along with several mixed-model upgrades and many other upgrades, fixes, and polishes. The two new features LD Score Computation and Binning and Compute GBLUP Using Bins, while they can… Read more »
Our software solutions and partners have brought dramatic improvements to the secondary and tertiary analysis stages of variant evaluation. Regarding secondary analysis, we’ve discussed increased efficiencies in speed and overall accuracy in the variant calling process with Sentieon. On the tertiary side, we have explored numerous workflows in VarSeq highlighting filtration to clinically relevant variants, as well as the automated… Read more »
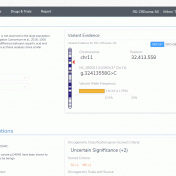
The detection and interpretation of Copy Number Variants (CNVs) is vital for the clinical evaluation of individuals with a wide range of disorders. Golden Helix has remained at the forefront of CNVs in Next-Gen Sequencing (NGS) data since 2016 with the release of VS-CNV, our solution that allows you to both detect and analyze CNVs directly from NGS data. Earlier… Read more »
It doesn’t take much effort to find articles discussing the value of Next-Generation Sequencing (NGS). There is a consistent tone amongst authors that implementing NGS pipelines are critical for clinical efficiency in both hereditary disorders and somatic. However, NGS strategies do not come without their own challenges. Challenges include not only the detection and calling of high quality/probability variants from… Read more »
Golden Helix has secured its reputation as a global leader in Next-Generation Sequencing solutions for over two decades. Today, we have been included in the esteemed Inc. 5000 list of rapidly growing American companies. Out of the 6 million business in the United States, Golden Helix has been honored with a spot in the annual list for the 2nd consecutive… Read more »
The COVID-19 Pandemic ferociously spread across the globe bringing our daily lives to a quick halt – desks, streets, and towns nearly empty as our world united together to help “flatten the curve”. While most of us were introduced to remote work, medical workers from all different specialties were called upon to help treat and test COVID-19 patients. One long… Read more »
Golden Helix software provides huge analytic gain in handling large-scale genomic data. For example, a number of VarSeq users run cohort projects of whole genome level data processing hundreds of millions of variants at a time. However, many of our users are running gene panel level data for custom panels related to cancer (both hereditary and somatic), autism, cardiac, and… Read more »
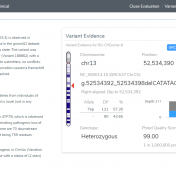
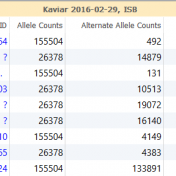
In the search for disease causing mutations it is important to determine if the variant has been previously observed in humans and at what frequency. With the advent of increasing genomic information, there is now a variety of different databases and annotation sources that can be utilized. For some, this could be a tedious task that leads only to implementing… Read more »
In this month’s Customer Publications blog post, our VarSeq software is taking center stage! From whole exome sequencing to copy number variant calling, VarSeq can be used for a range of scientific investigations. Although this blog features several examples of cancer investigations in human patients, it’s interesting to see how this platform can be utilized in a variety of investigational… Read more »
It is common knowledge that variants can be germline or somatic depending on whether the variant was inherited or acquired after birth. A well-known example is cancer-causing mutations in the BRCA genes, wherein the mutation may or may not have been inherited. Understanding the origin of the cancer-causing mutation is important when assessing potential treatment options as well as identifying… Read more »
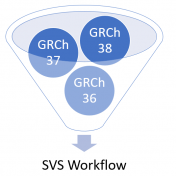
The world has been making a shift to use GRCh38 human genome reference coordinates, but the transition has not been fast. Many of the mainstay human catalog projects are changing to use native GRCh38 catalogs, or are remapping their current data to GRCh38 coordinates. While this seems to be the advancing goal, it is leaving researchers and analysts with the… Read more »
Thank you to everyone who joined our webcast, “Whole Genome Trait Association in SVS.” If you missed the live event and are interested in knowing what we talked about, you may access the recorded event below: Our Live Q&A generated a lot of great questions. Unfortunately, we were unable to answer them all, but we have compiled some of the… Read more »