In many cases, VarSeq users typically run single trio projects or perhaps an extended family project. Not only are all the inheritance model algorithms available in the VarSeq software to capture de novo, dominant, or recessively inherited variants but there are a number of quality control fields to help ensure the pedigree was set up properly. The last thing any… Read more »
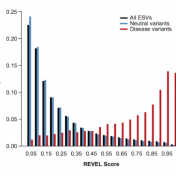
Typically, researchers are looking for rare variants in their next generation sequencing datasets. However, most of the nonsynonymous variants have unknown significance because there is an inherent difficulty in validating large numbers of rare variants or even detecting rare variants with high statistical power. In lieu of this issue, computational tools are needed as they accurately predict the pathogenicity of… Read more »
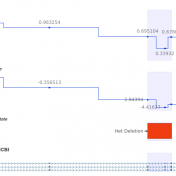
In trio workflows, one of the most important factors in scoring a variant is understanding how that variant is inherited from the parents. Likewise, when looking at extended families, the segregation, or presence of the variant among the affected versus unaffected individuals provides evidence for its pathogenicity for a given phenotype or disease. Given the nature of Copy Number Variants… Read more »
Golden Helix ships a variety of templates that are designed to provide a starting point for users to evaluate variants in VarSeq. Naturally, as users become more familiar with the software, there is a desire and necessity to tailor the template design to accommodate a more thorough variant analysis. To add to these template customizations there are several algorithms and… Read more »
Although VarSeq is intentionally designed to be a clinical NGS pipeline tool able to run a handful or even single samples through, we have many users who run large cohort style studies with the tool as well. One common use is to compare case/control data to isolate variants shared among affected individuals and exclude those in unaffected. One incredibly powerful… Read more »