Advances in high-throughput sequencing have allowed us to be able to detect structural variants such as copy number variants in addition to small variants such as SNVs and indels. We provide users with an industry-leading CNV calling algorithm to detect CNVs directly from their next-generation sequencing data including whole genome, whole exome, and gene panel datasets, and also import CNVs… Read more »
The overall objective of Trio Analysis is to leverage inheritance data to determine the ancestry of variants in the proband, whether that be through transmission from the mother and father, or of de novo origin. We have previously covered family based analysis in this blog post and this webcast, but both of these sources have been based on variants brought… Read more »
While the interpretation of germline variants generally focuses on the pathogenicity of a variant for a specific disease, the interpretation of somatic variants is centered around each variant’s impact on clinical care. As a result, clinical trials play an important role in assessing the clinical significance of somatic biomarkers, with the AMP Guidelines assigning a higher level of evidence to… Read more »
Here at Golden Helix, we’ve sought to develop top-quality bioinformatic software to handle not only high-throughput clinical next-gen sequencing pipelines but also cater to research groups exploring their cohorts. Our tools fit in this NGS pipeline by importing the VCF and BAM files for downstream filtration, annotation, and classification/interpretation as well as clinical reporting. It was obvious early on and… Read more »
Curated databases are a real time saver when compiling published evidence to support your variant evaluations and classifications. Leveraging the curated databases at your fingertips in our VSClinical variant interpretation hub is even more efficient. Not only does VSClinical provides users with automated variant classification for germline variants according to the ACMG guidelines and somatic variants according to the AMP… Read more »
An under-utilized use of VarSeq is the ability of mining raw variant data in GenomeBrowse for relevant literature. By bringing in various public and private annotation sources, GenomeBrowse allows the user to interface with raw variant data in a compressed and manageable view. This blog will show you how to leverage these sources to power up your search for variant… Read more »
Although best known for its auto-generation of custom reports, VarSeq comes with a slew of options for exporting your data. In this blog, we will review some of the lesser-known methods for exporting your data into usable formats. These four export options can all be found under the Export tab at the upper left corner of your VarSeq interface (Figure… Read more »
Global population frequency catalogs like 1kG Phase 3, gnomAD, DGV, and others are excellent resources for identifying rare variants in your copy number variant (CNV) analysis. However, they are not exhaustive, and the reality is a lot of variants that are missing from global population frequency catalogs are still common variants. At the same time, CNVs that are identified by… Read more »
In this blog post, I am very excited to talk about The Broad Institute’s release of the latest version of gnomAD, v 3.1.2, which is now available for use as an annotation source in your SVS or VarSeq projects. For VarSeq users, I also want to point out that gnomAD v3.1.2 can also be used as a population frequency in… Read more »
As the number of genes on a gene panel increases, there is the possibility of picking up variants of medical significance that are not related to the primary indication for the test. Especially with large gene panels, exomes, and genomes, it is medically and ethically important to report variants that may be actionable to the patient. These include variants implicating… Read more »
When it comes to clinical variant analysis for germline variants using ACMG guidelines, we understand that the clinical report is essentially the receipt for your services for the patient. In our recent webcast, Advanced Report Customization in VSClinical, we displayed several examples of report templates to show off the range of possibilities our users have to format their clinical report…. Read more »
We are very excited to announce that just last week, we released VarSeq 2.2.4! In the past few months, we have been building the excitement for the 2.2.4 version of VarSeq with several webcasts in which we describe some of the headlining features in detail such as the new support for Gene Panels and Gene Lists, PhoRank Clinical, and Customized… Read more »
Thank you to everyone who attended Using Golden Helix CancerKB to Accelerate NGS Cancer Testing! I had a great time showing off Golden Helix CancerKB and how it can enhance NGS analysis in cancer, I certainly hope you enjoyed it! If you were unable to make the live session, we have added the on-demand recording to our site, or we… Read more »
As a newer member of the Golden Helix FAS Team I, like many of our customers, went through a phase of learning the ins, outs, and shortcuts of clicking around the program. In an effort to expedite new users coming up to speed, or to teach some tricks to experienced users, I’ve compiled a list of the top things I… Read more »
The VarSeq CNV calling algorithm, VS-CNV, is a powerful tool for calling CNVs from the NGS coverage data stored in your BAM files. However, before this algorithm can be deployed in a clinical setting, it must be tuned and validated using data that is representative of your lab’s NGS workflow. In the past, this validation process could be difficult, as… Read more »
As a lab or group scales the number of NGS samples analyzed, it is important to automate the sample analysis pipeline from the sequencer to the point where it is ready for a variant scientist or lab personnel to follow the interpretation workflow and draft a clinical report. VSPipeline leverages the core VarSeq capability to create reproducible test-specific workflows through… Read more »
The customer-published articles this October cite the range of Golden Helix’s VarSeq annotation range and capability. The following publications feature everything from annotating an Italian nobleman mummy, assisting in identifying mutations in primary congenital and juvenile glaucoma, new mutations associated with muscular dystrophinopathy, and germline variants associated with head and neck cancer. In each of these cases, VarSeq was utilized… Read more »
Recently we have released blog posts discussing updates to annotations in VarSeq such as ClinVar and COSMIC. Keeping with that trend, in this blog post, I will discuss the most recent updates to the Golden Helix CancerKB database. For those how may be unfamiliar with the Golden Helix CancerKB source, it is a professional curated set of interpretations for the… Read more »
Golden Helix VSClinical provides a guided workflow interface for following the ACMG and AMP guidelines to evaluate variants and CNVs for NGS tests. The output of this work is most often a lab-specific clinical report. Since it was introduced, we have provided a powerful Word-based templating system to allow labs the ability to generate customized reports to include specific content… Read more »
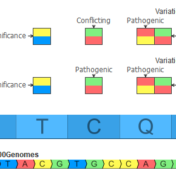
In the September 2021 monthly update to our curated ClinVar track, we made some changes that will result in roughly another 7,000 Likely Pathogenic and Pathogenic variants being available for annotation and use in the ACMG auto-classification system. Consensus Between Labs ClinVar has nearly one million unique variant classification records that are curated into multiple annotation tracks used in VarSeq and VSClinical on a monthly basis. Clinical… Read more »