Golden Helix is excited to announce a new round of novel and updated annotations; including a frequency track, a region track, and a gene track. All three of these tracks were created with the use of VarSeq and its Convert Wizard functionality. First, the expansive 1000 genomes track (1kG) has been updated to include sub-population allele frequencies and heterozygous and homozygous sub-population counts. Also, Golden Helix now provides a region track with data taken from the Online Mendelian Inheritance in Man (OMIM) catalog to help support the recent CNV caller capability in both VarSeq and SVS suite product lines, as well as an updated Ensembl gene tracks for human genome GRCh 37 and GRCh 38 genome sequences.
New 1000 Genomes Genotypes and Sub-Population Frequencies
The 1000 Genomes (1kG) catalog has been a common reference source due to its expanse of over 85 million variants collected from over 2500 individuals. With the newly released 1kG annotation track, we wanted to expand the utility of this popular source by adding sub-population allele frequencies, as well as allele and genotype counts.
The available annotation fields in the previous 1kG track are shown in the list below and come directly from the data provided by the 1000 Genomes consortium. This track did offer the alternate allele frequencies for sub-populations, but no sub-population allele counts or specific het/hom genotype break-out values.
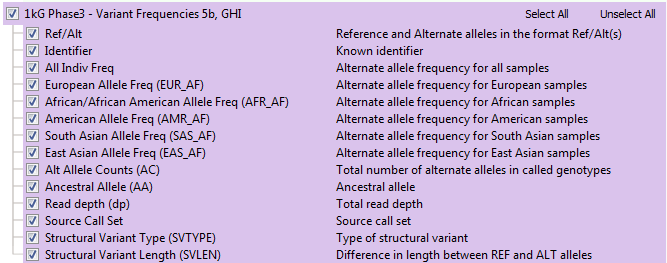
Note: for the field acronyms, the sub-population labels are described below.
- AMR-Latino (mixed American)
- AFR-African
- SAS-South Asian
- EUR-European
- EAS-Eastern Asian
Now, the updated 1kG track fields are shown below. Here, each subpopulation has an alternate allele counts field (Allele Counts), an Allele Frequencies field (Allele Frequencies), a total number of observed
alleles (# Alleles), a count of the number of heterozygous genotypes (# Het), and a count of the number of homozygous or hemizygous genotypes (# HomoVar).
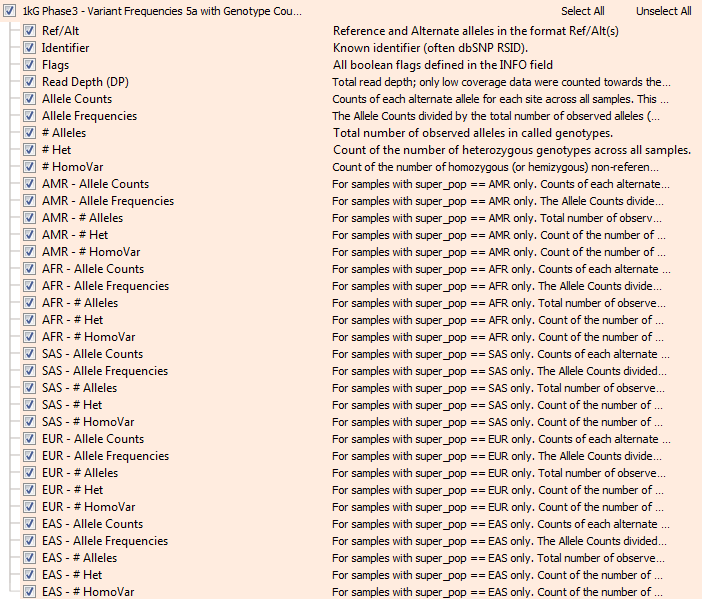
This new frequency track allows for more precise interpretation of rare variants in an ethnically matched manner. ExAC and gnomAD provide genotype counts and shown the value of being able to know whether a rare variant ever occurs in a Homozygous state or is only ever in a carrier (Heterozygous state). As a side-note, we computed these counts using VarSeq’s own Allele Counts algorithm run on the full genotype data provided by the 1000 Genomes consortium
OMIM Regions
Golden Helix has offered OMIM genes and phenotypes annotation tracks but now offers an OMIM regions track. While many OMIM entries are specific to a gene, some ore more broadly defined as diseases associated to genomic regions and sometimes to specific copy number states of that region (Gains vs Losses).
This track can be used in conjunction with the new VarSeq CNV annotation capabilities because it provides a CNV “Type” field which will match the annotation records to the type CNV events called in your NGS samples using VS-CNV.
Each event provides the relevant OMIM entry information, including PubMed literature information and phenotype descriptions. The annotation track fields are displayed below.
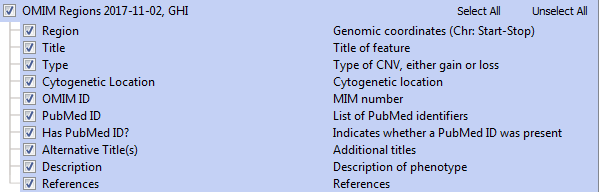
Note that Golden Helix provides OMIM as a premium annotation source and accessing OMIM Regions requires the inclusion of OMIM in your license package. Please contact us if you are interested in adding this to your existing license.
Updated Ensembl Genes
The Ensembl Gene tracks have been updated this year to version 87 for the GRCh 37 genome and to version 90 for GRCh 38 genome. The first of three annotation tables that will be created when the Ensembl annotation is applied shows the gene annotation name, effect, and clinical relevance.
Although many clinical labs in the US use the NCBI RefSeq Genes based transcript and gene set for clinical interpretation so they can report their variants in HGVS using NCBI identifiers, European labs often use the Ensembl based transcript and gene set provided in this track.
VarSeq is extremely flexible in your choice of gene annotations, and you can use either or both gene sources in your analysis.
Here is a review of the fields the VarSeq transcript annotation provides. The first set of fields are “Summary” fields provided for each variant:

The second set of columns provide details on the interaction between each transcript and a given variant in VarSeq. If you drill into the details of a variant, you may find there is a one-to-many relationship between a variant and the transcripts it overlaps. While the summary field is great for filtering and picking out the one Clinically Relevant transcript and HGVS you may want to report, you can view the other transcript interactions for more details and review the impact of this variant in other transcripts.
Other than the Transcript names and of course differences in the gene sets themselves, these two tables will have the same information regardless of whether you are using NCBI’s RefSeq Gene or Ensembl Gene for your gene annotations.
Note the field names are shown below with the default display categories checked, but you can change the visibility or click “Show Hidden Fields” in the Details pane to dig deeper!

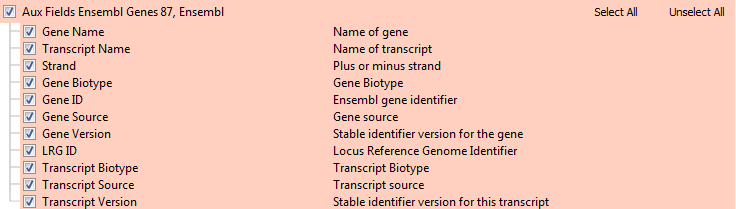
Finally, other than defining the transcript and gene definitions themselves, our gene annotation sources provide other meta-data about the gene such as their RNA type as well as various database references. The Auxiliary Fields table provides these per-source fields and they are quite different between RefSeq and Ensembl based on what is provided in their respective primary sources.
Golden Helix is proud to offer these new and updated annotation tracks to our customers to help analyze and crack the genetic code.