ClinVar is the NCBI variant database that focuses on the categorizing of variant alleles and their interpretation from a clinical standpoint. This has made it a great resource, especially for those seeking variant allele disease correlations and pathogenicity. And this all worked fairly well, but it was changed…
Previously, the ClinVar variant track annotation took some time to curate due to the fact that the data was provided from the ClinVar website in multiple files consisting of multiple file types (this has been outlined in the previous Golden Helix blog posts that can be found here and here). Golden Helix curated ClinVar formerly using the variant definitions for variants with dbSNP identifiers VCF file format and adding the remaining variants found in the plain text file. The VCF file required further transformations, as it contained just one record per variant (that is to say, one record providing the chromosome position, and reference and alternate allele information), but more information could be gleaned from this data. Working backward from the VCF file, a per-disease association could also be extracted to improve the usefulness of the ClinVar records. The picture below shows vertical stacks of variant records where multiple diseases are associated with that variant.
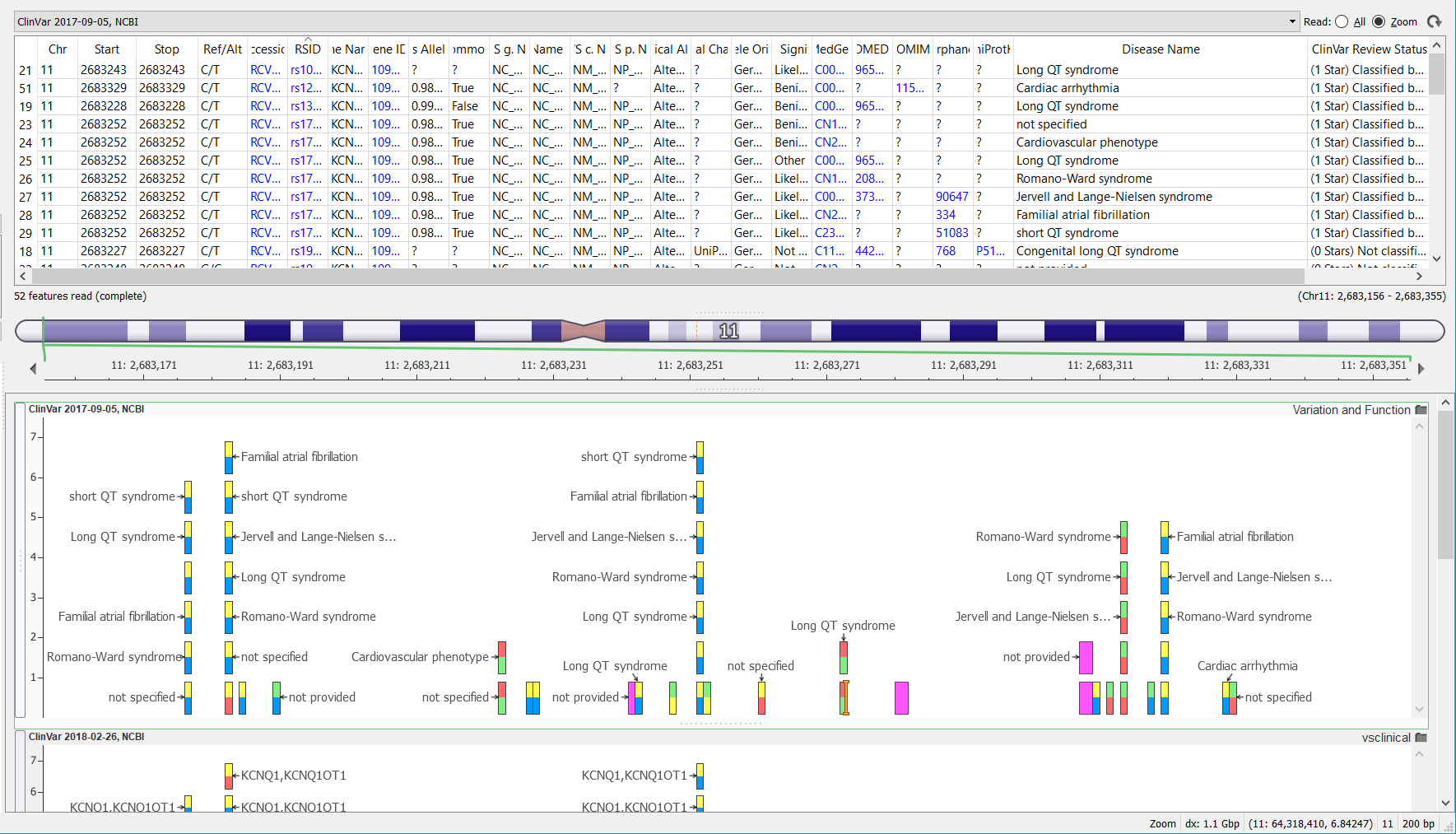
The ClinVar team has known about the limitations of the VCF file (only containing variants already in dbSNP) for a while and the resulting complexity for variant curators like ourselves. So they have finally decided to change their tactics.
ClinVar gets a makeover
This year marks an update to the ClinVar methodology. Now, the updated VCF file includes all the variants and no longer relies on the dbSNP VCF writer. They also compacted the annotation fields so that all the previous information was collapsed into one variant entry. One of the multiple entry variants is shown below with one entry on the top from the new VCF file format and the previous style with multiple entries shown on the bottom.
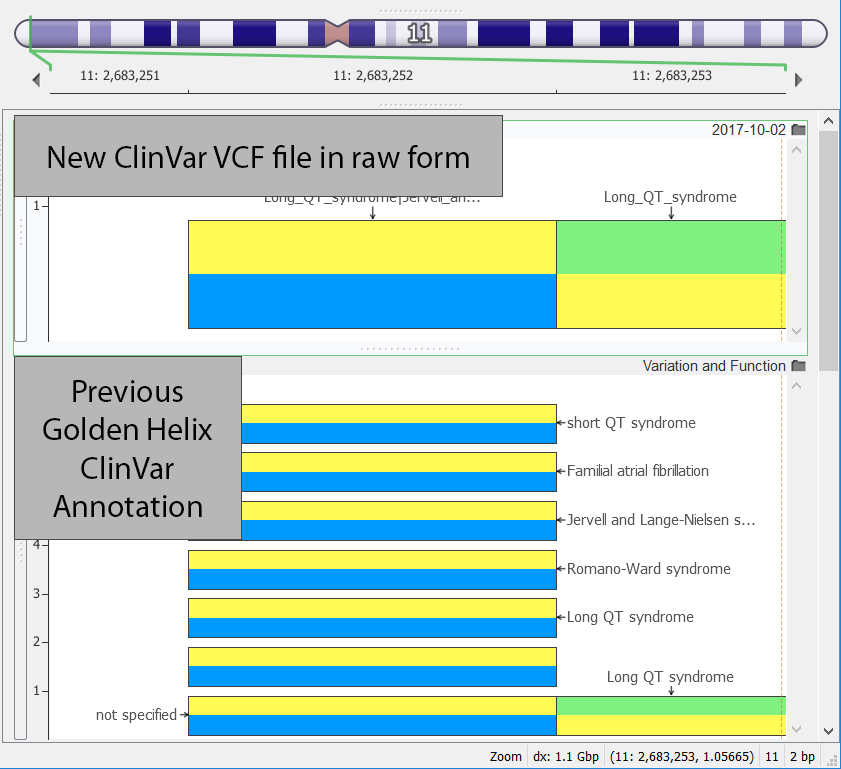
On the positive side for annotating, though, is that the new VCF track included a new concept of a variant level classification/curation status. This can be seen below in the documentation output in VarSeq below where the 16th and 17th row show the Clinical Significance at the Variant Level and the Review Status at the Variant Level, respectively.

The field in the documentation image above titled CLNDN is the disease name field and collapses all the records into one long string. This VCF file is a great improvement to the jumble of files we dealt with earlier, but the redone VCF loses the fidelity of knowing what’s going on at the per-disease level. We are no longer able to “work backward” to the disease level variant records like we could previously and clearly correlate the clinical entries to the corresponding disease, but there is a solution.
The XML Solution
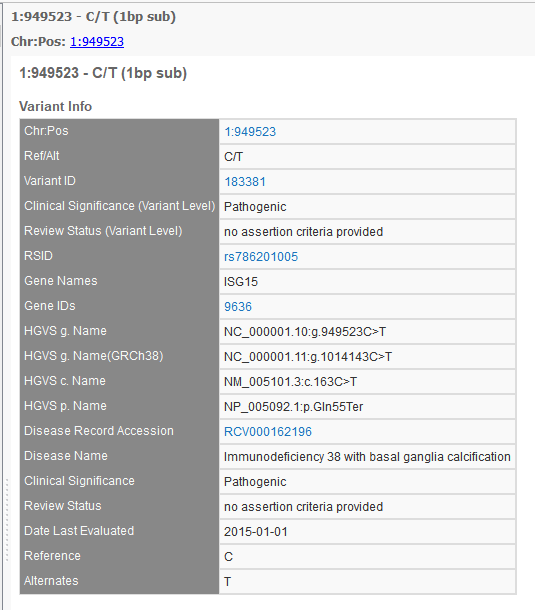
So, we have been in discussions with ClinVar folks, and they recommend we take a look at the new variant-level XML dump, which matches very closely to the ClinVar variant webpage as well as provides details on the per-disease assertions. Whereas before, researchers annotating had to use the accession number (or RCV number) to track down a variant in the ClinVar database, now a Variant ID can be used to define the record that holds or links all the information for that particular variant-a variant overview snapshot. The perfect solution. And an example of one such variant webpage on the ClinVar website is shown below.
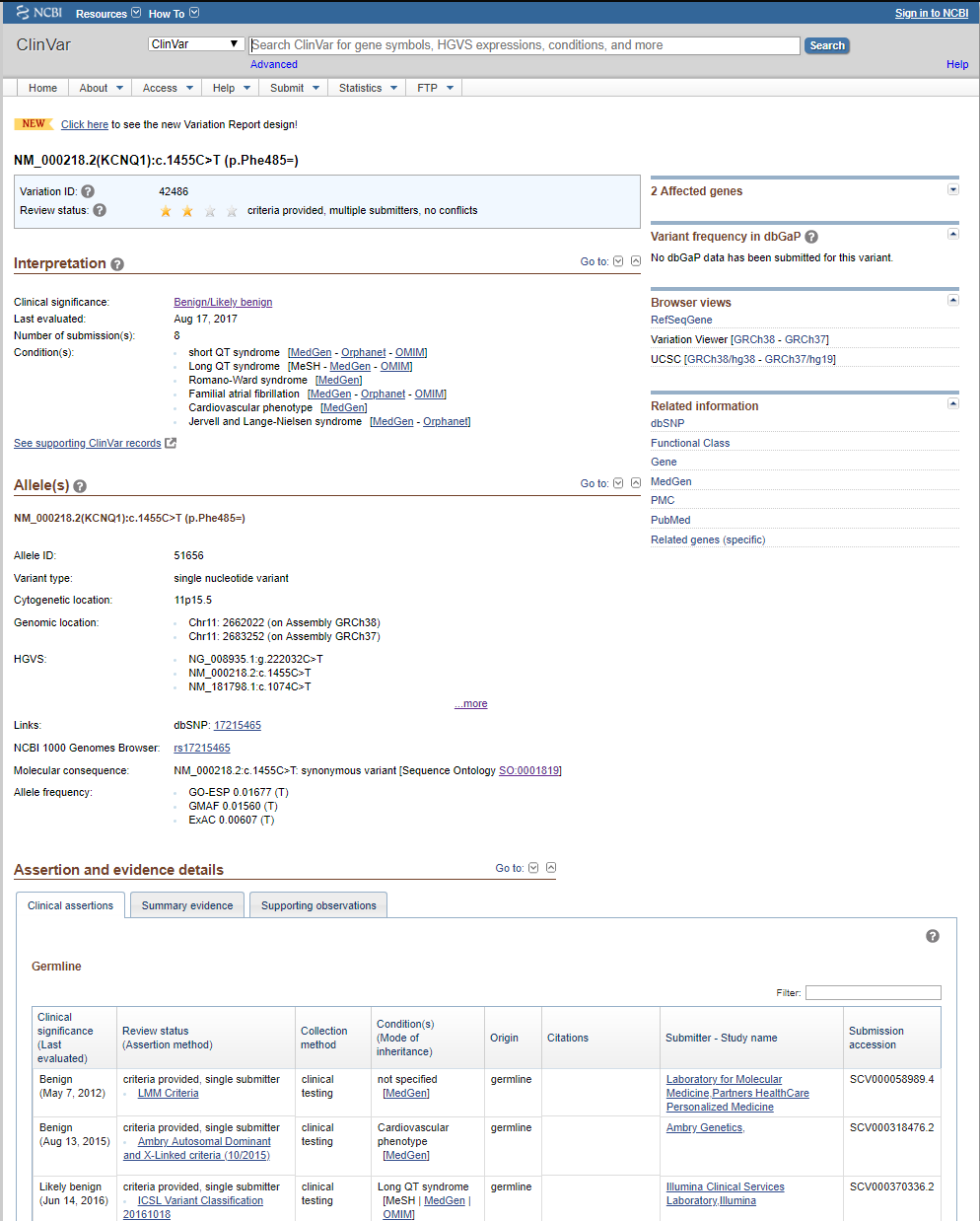
This new XML report has been in beta and seen some changes since late last year but has now stabilized and we are fully switching our curation over to it.
The added benefits include the variant level pathogenicity/review, as well as the artifacts of the older necessary annotation style (VCF+ TXT) merging, are gone. We also took this opportunity to shave off some of the old cruft that was not really helping with variant annotation. The new variant track does not include the following database identifier numbers: 1000Genomes Allele frequencies, MedGen, SNOMED_CT, OMIM, Orphanet, UniProtKB, but does add the helpful fields of Variant ID, Clinical Significance at the Variant Level, Review Status at the Variant Level, and Date Last Evaluated. Also, the disease accession number does not track the decimal place version quantifier like in the Old version, so a link to outdated versions will be avoided. This is reiterated by the newly included ‘Date Last Evaluated’ field which corresponds to the version date/date last evaluated.
The new XML file also does not include the Citations field, but this information was included in more detail in the new XML file, so this information was split into an entirely separate track called, ClinVar Assessments (which we are working on and will be released shortly). A comparison between the older documentation and the new documentation taken from the new XML file is shown below.
| Old ClinVar release documentation in VarSeq | New XML sourced documentation in VarSeq |
The new track does away with:
|
The new track adds:
|
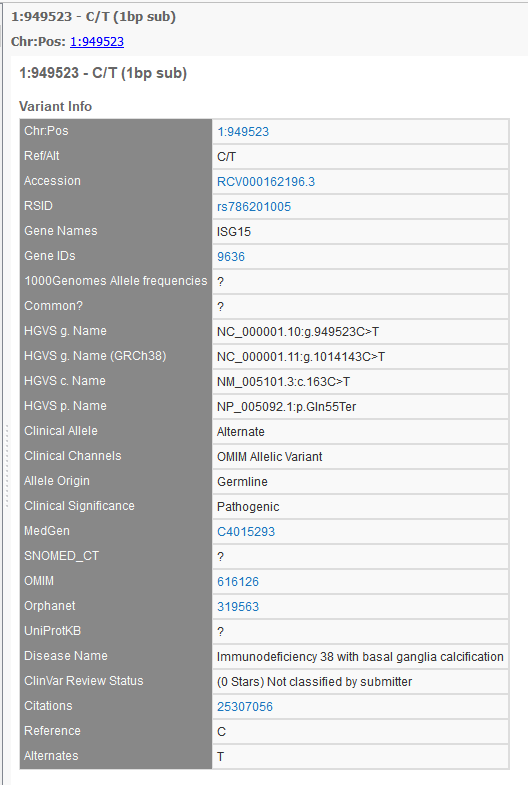
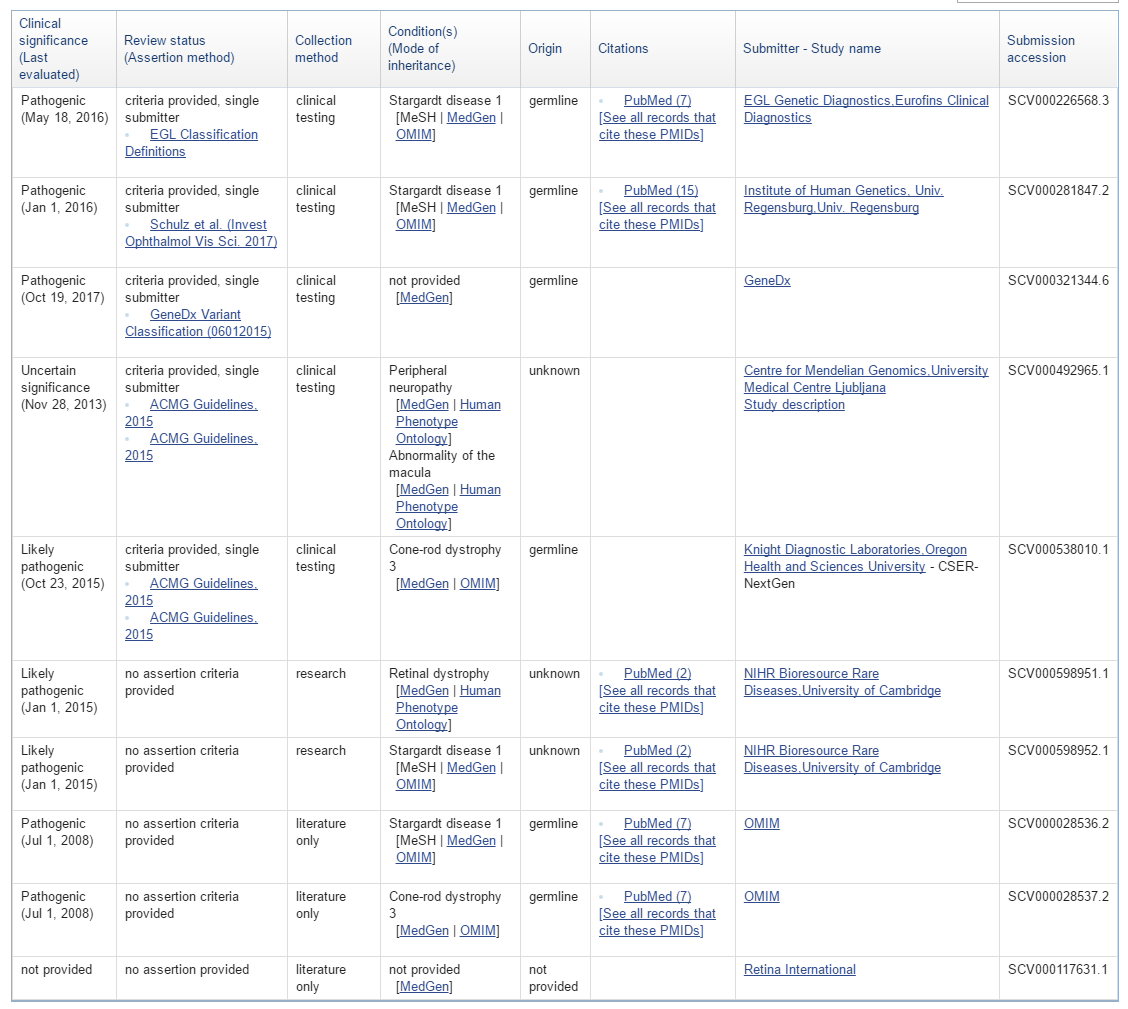
Part of the Clinical Significance at the Variant Level is the new concept of a “conflicting interpretations” status. An example of a variant entry on the ClinVar website is shown below with multiple clinical significance entries and dates.
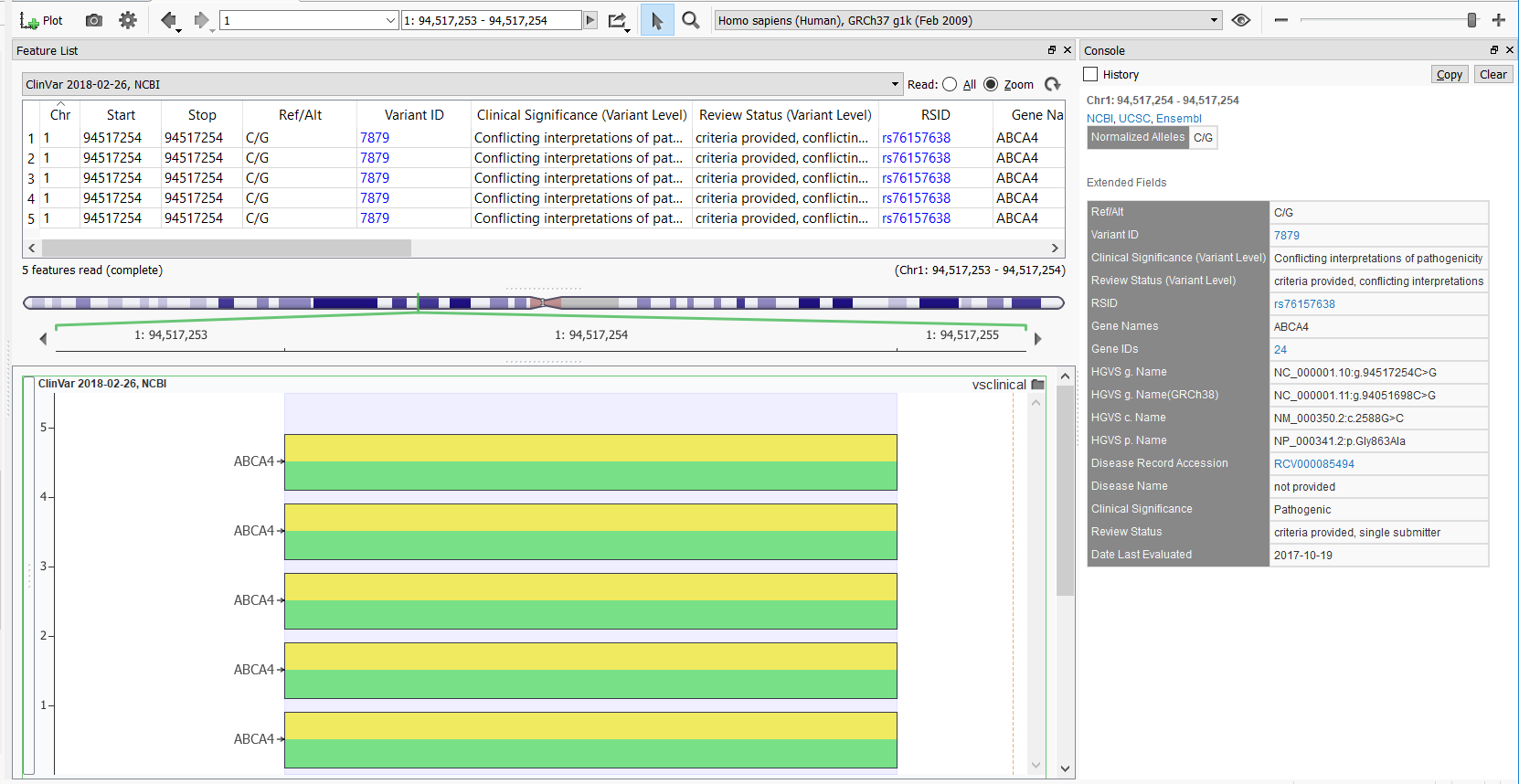
This particular variant produces multiple variant entries in the GenomeBrowse variant viewer, but the newly added Variant ID field now provides a hyperlink to the above webpage and the Date Last Evaluated field now easily shows the most recent date at which that entry data was evaluated in the ClinVar database. The GenomeBrowse entry with documentation for the variant above is shown below.
Overall, we are very happy with this new database release. It provides a cleaner and more concise reflection of the ClinVar database that matches their website and makes for a better analytical workflow. The new ClinVar track makes it easier to filter to the variants that you’re confident in prioritizing in your analysis and is overall more efficient. Be sure to check it out!