Our latest release of the VarSeq software has had a major upgrade with the addition of the new CNV ACMG guidelines! Here are some recent webcasts we’ve given covering the new guideline tool:
Family-Based Workflows in VarSeq and VSClinical
A User’s Perspective: ACMG Guidelines for CNVs in VSClinical
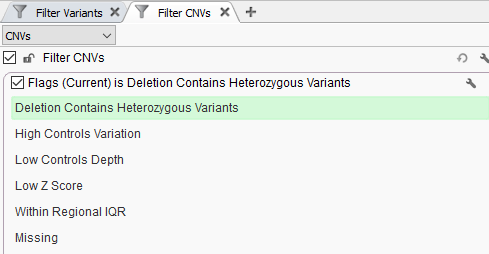
Not only does VarSeq 2.2.2 come with the new guideline tool, but also a number of new features related to CNV analysis. Any variant analysis is going to include the capability of excluding low-quality calls or artifacts. The CNV workflow in VarSeq has had this quality flag assessment since the introduction of our CNV computation. However, we added a new field to make this quality assessment even more thorough. Figure 1 shows our latest quality flag “Deletion Contains Heterozygous Variants”, and it works just like it sounds, to isolate full deletion events that overlap with heterozygous variants from the VCF. In some cases, this may be a deletion event that could be a false positive due to low-quality coverage for the sample when compared to the reference sample set, or perhaps the variants in the VCF are false-positive SNPs or indels that need to be ignored.
Additionally, we’ve optimized the karyotype values listed in the CNV table to show up only when an event is at least as large as one cytoband region. We’ve also added new features to the CNV caller menu. As seen in Figure 3, when setting up the CNV caller, click the advanced tab and look toward the bottom to see these new options.
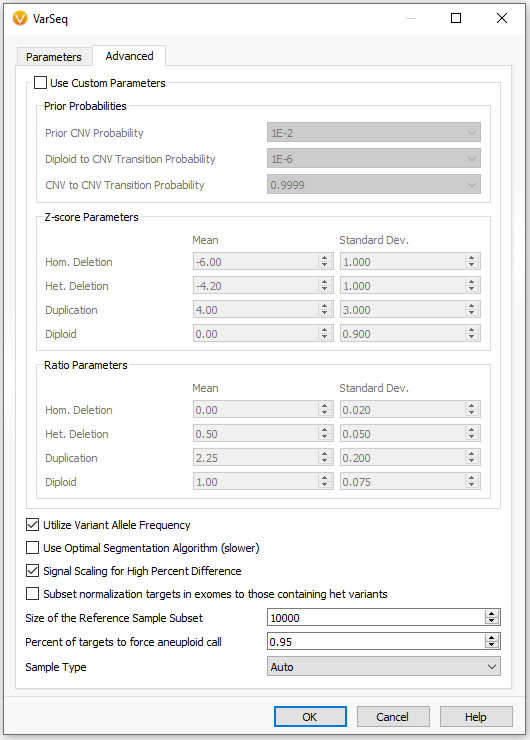
One new option is to modify the size of the reference sample subset. The main reason for this addition is that we’ve had some users in the past collect a massive number of reference samples. This was problematic because the computation took an unreasonable amount of time to complete so we implemented a backend feature to subset the samples for a smoother run time and experience. However, some users still benefited from a wider range of reference sample subsets, so we added a modifier where the user can manually set their preferred subset size. Other example advanced options include:
- enabling/disabling signal scaling for high percent differences compared to reference samples.
- modifying the percent of region targets forcing any aneuploidy calls.
- selection of data or sample type as Gene Panel or Exome to allow karyotype calls for large whole chromosome events.
- subsetting normalization targets to exomes with heterozygous variants ideal for highly mutated tumor samples.
The evolution of these CNV tools and output has always been centered around the need for resolving customers’ issues. Each pipeline is unique and requires a necessary understanding of important issues such as accurate target definition and management of low-quality coverage. Not only can users find a breakdown of the new options and output in the user manual linked above, but are also welcomed to reach out to info@goldenhelix.com if you would like more formal training or to investigate any nuances with your VCF and BAM data. Feel free to also check out some of our other blogs that always contain important news and updates for the next-gen sequencing community.