In recent months we have been updating our public annotation library to include the most recent versions of existing sources, as well as include new sources. All of these annotation sources are compatible with our three major products, VarSeq, SVS, and GenomeBrowse, and can be used for visualization, annotation, and filtering.
dbNSFP NS Functional Predictions 2.8, GHI and dbNSFP Predictions 2.8, GHI
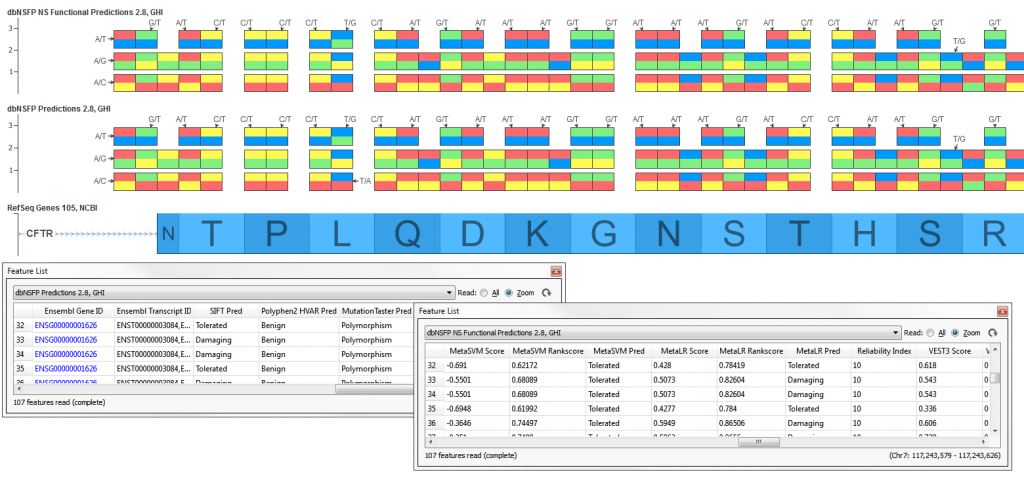
The full dbNSFP NS Functional Predictions 2.8, GHI is an integrated database of functional annotations from multiple sources for the comprehensive collection of human non-synonymous SNPs (NSs). Its current version includes a total of 87,347,043 NSs and 2,270,742 splice site variants. It compiles prediction scores from nine prediction algorithms (SIFT, Polyphen2, LRT, MutationTaster, MutationAssessor, FATHMM, MetaLR, MetaSVM, VEST), eight conservation scores (PhyloP 46way Primate, PhyloP 46way Placental, PhyloP 100way Vertebrate, PhastCons 46way Primate, PhastCons 46way Placental, PhastCons 100way Vertebrate, GERP++ and SiPhy) and other function annotations.
When it is not feasible to download the full track (~5.6 GB in size) a subset track is available, dbNSFP Predictions 2.8, GHI which contains only the 5 predictions for SIFT, Polyphen HumVar (HVAR), MutationTaster, MutationAssessor, and FATHMM (~439 MB in size).
These tracks are available for the three major human genome builds; NCBI36 (hg18), GRCh37 (hg19) and GRCh38 (hg38).
dbscSNV Slice Altering Predictions 2014-11-09, GHI
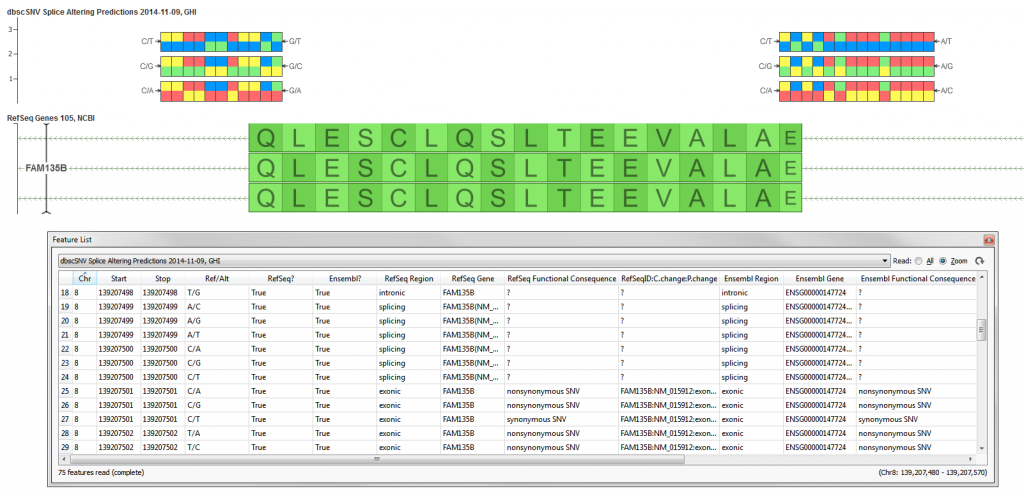
dbscSNV is an attached database that is available alongside the dbNSFP database and includes all potential human SNVs within splicing consensus regions (−3 to +8 at the 5’ splice site and −12 to +2 at the 3’ splice site), i.e. scSNVs, related functional annotations and two ensemble prediction scores for predicting their potential of altering splicing. The scores are computed by running various splice site prediction algorithms (MaxEnt, SplitPort, etc.) and then using a machine learning technique of AdaBoost or Recursive Partition trees (ada_score, rf_score) to provide an “ensembl” prediction. The algorithms used look at the sequence of the site + flanking region to predict computationally whether a splice site may exist there. The alternate sequence is compared to the reference and the score is descriptive of “disrupting” the naturally occurring splice site or introducing a new one. Please see the method paper for the source of the data for further details.
This track is available for the GRCh37 (hg19) human genome build.
Related Article: Revisiting the Five Splice Site Algorithms used in Clinical Genetics
ClinVar 2015-01-06, NCBI and ClinVar CNVs and Large Variants 2015-01-09, NCBI
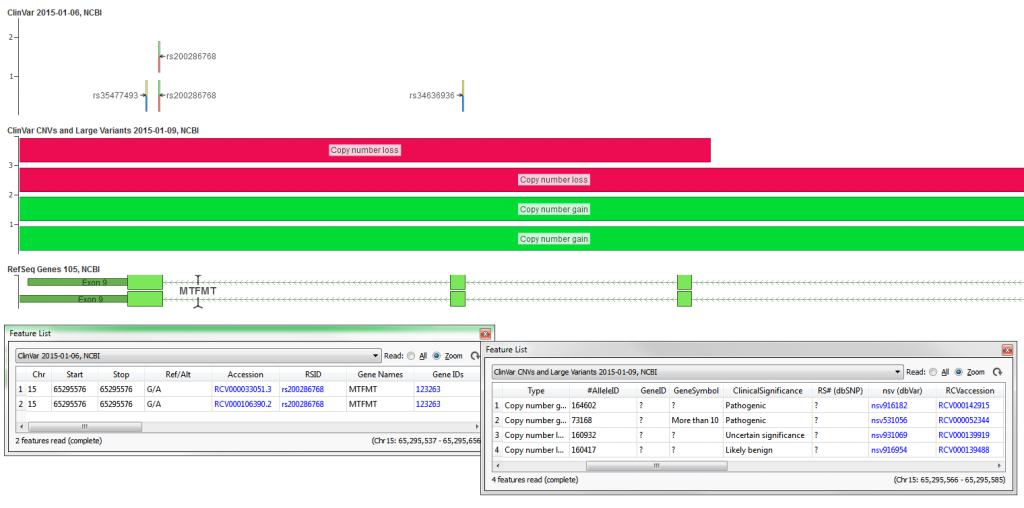
The ClinVar database from NCBI is a public reports archive of the relationships among human variations and phenotypes, with supporting evidence. The ClinVar supplies the data in several formats, the VCF format is provided in a “one record per Ref/Alts pairs” convention and thus places multiple ClinVar records on a single line in the VCF file. To create the ClinVar 2015-01-06, NCBI track these lines were split into their own records and their values were formatted into useful representations. For example, rs200286768 has 2 records in ClinVar, each with a clinical classification in regards to a different disease (see Figure 3). This track will show each record independently at that same genomic location. The variants were also left-aligned (copying records to left-most position when they are not there already) to support annotation against left-aligned NGS data.

ClinVar also provides a delimited text file with the records from their database. From this delimited file the ClinVar CNVs and Large Variants 2015-01-09, NCBI track was created. The track contains all variants in the ClinVar database with a length greater than 200bp.
These tracks are available for the two most recent human genome builds; GRCh37 (hg19) and GRCh38 (hg38).
COSMIC Mutations Left Aligned 71, GHI and COSMIC Cancer Gene Census 71, GHI
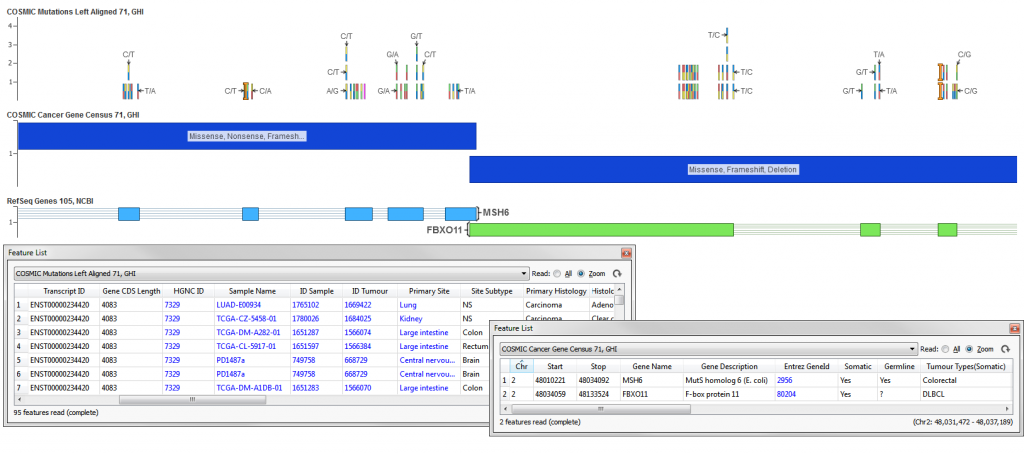
The COSMIC Mutations Left Aligned 71, GHI track contains data from the Catalogue of Somatic Mutations in Cancer (COSMIC), combining curation of the scientific literature with tumor resequencing data from the Cancer Genome Project at the Sanger Institute, U.K. The track was generated from a CSV file which contains all samples analyzed for every gene in COSMIC found with/without mutation. The COSMIC mutation data only provides allelic information in HGVS notation (CDot). A custom script was run to derive, using the CDot and the reference sequence at the provided genomic coordinates, the Ref/Alt pairs for a given site and to split records into individual Ref/Alt/Sample records. The variants were also left-aligned (copying records to left-most position when they are not there already) to support annotation against left-aligned NGS data.
The COSMIC Cancer Gene Census 71, GHI track contains a list of cancer census genes from the COSMIC cancer gene census. The cancer gene census is an ongoing effort to catalog those genes for which mutations have been causally implicated in cancer.
These tracks are available for the GRCh37 (g1k) human genome build
Related Post: New COSMIC Database for NGS Cancer Analyses
Downloading the tracks and requesting new ones!
To obtain all of these new and updated tracks through SVS and VarSeq, go to Tools > Manage Data Sources and select them for download through the Public Annotations repository. For GenomeBrowse the Public Annotations repository can be found by going to File > Add to open the Data Source Library. Once downloaded the tracks will be available in your local annotations folder which is the default location (…/Golden Helix/Common Data/Annotations) for all three Golden Helix products.
We are continuing to add new annotation tracks, support new species, and update genome builds. If you would like to request a particular database be converted into an annotation track or would like to see a particular species or build be available in SVS and GenomeBrowse please email us and let us know!
Pingback: New and Updated Annotation Tracks Now Available! | Our 2 SNPs…® | Raony Guimarães
Pingback: VarSeq is a better ANNOVAR, snpEff and VEP | Our 2 SNPs…®