The Genome Aggregation Database (gnomAD) is a resource developed by an international coalition of investigators, with the goal of aggregating and harmonizing both exome and genome sequencing data from a wide variety of large-scale sequencing projects (1). We have covered this annotation in-depth in other blog posts, but this resource contains over 125,000 exome sequences and around 16,000 whole genome sequences, which can be utilized to filter down variants using allele frequency and other fields to identify clinically relevant variants.
The online gnomAD database provides users the ability to look up allele frequency for a given variant and will display Allele Count (AC), Allele Number (AN), and Allele Frequency (AF) across exomes, genomes, and the combination of the two (Figure 1). Golden Helix also provides this information, but it is broken down into individual tracks: one for gnomAD exomes and one for gnomAD genomes. As requested by multiple users, we are going to provide a merged gnomAD exomes and genomes annotation source that can be loaded into projects and integrated into filter logics to filter on the total gnomAD exomes and genomes allele frequencies!
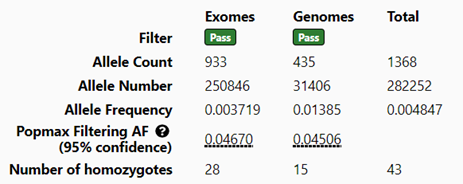
How was the merge was accomplished?
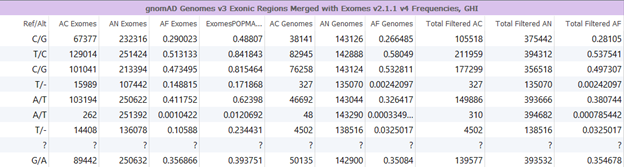
The development team at Golden Helix has curated this database for both GRCh37 and GRCh38. To create these databases, RefSeq genes +/- 20 base pairs was used to subset the gnomAD Genomes annotation to exonic regions and then it was merged with gnomAD exomes. To curate the GRCh38 version of this annotation there was an extra liftover step. To maintain consistency, the total AC/AN and homozygous count were kept for each source, as well as the POPMAX field for gnomAD exomes. The FILTER fields were kept for each source, and when they were value PASS, the counts were ignored in the computation of a Total AC/AN and AF field. This prevented inflated data, as they could potentially be counted twice for both exomes and genomes.
This annotation is available on the Staging Annotations server which can be accessed by opening up the Data Source Manager and selecting the small plus icon in the top left-hand corner. Then, for Location Type, select Remote, and choose Staging Annotations from the dropdown menu for Location. Your settings should match those seen in figure 2.

The sources hosted on the staging often come with some caveats for use that we like to make users aware of. For this merged gnomAD source, not all fields that are available in gnomAD Genomes and gnomAD Exomes separately were used to merge these tracks. Also, as eluded to earlier, the FILTER field was used in calculation of allele count, allele frequency and allele number so naturally, some variants are dropped.
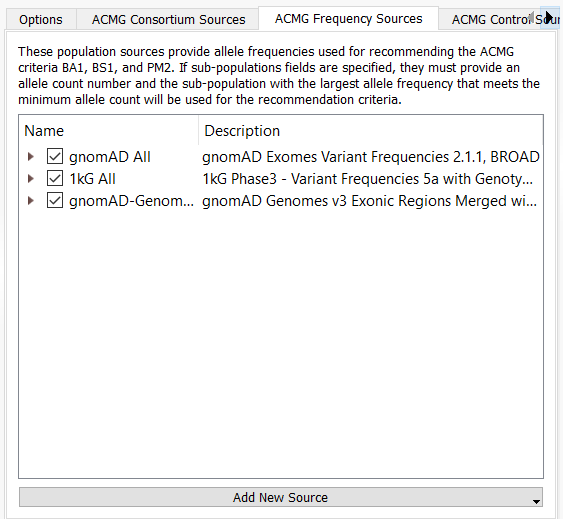
That being said, these databases can be loaded into the variant table and subsequently integrated into the filter logic. Additionally, you can optionally add this merged frequency database as an ACMG custom frequency source for use in VSClinical (Figure 3).
Altogether, we are happy to announce the upcoming release of the merged exomes and genomes gnomAD annotation track, which will be available for both GRCh37 hg19 and GRCh38 hg38. There are obvious utilities of this track, so we are very grateful for the customer feedback that prompted its development. If there are any questions about this post or the annotation itself, feel free to leave a comment in the section below! Interested in seeing a demo or trialing the software? Click on the button below to request a VarSeq eval: