Big data is here, but fear not, you don’t need a Hadoop cluster to analyze your genomes or your cohorts of tens of thousands of samples!
It turns out, for the kind of algorithms employed in variant annotation and filtering, running optimized local programs is often faster anyway.
As we support our diverse customer base, we have definitely seen the upward trend of sequencing whole genomes versus just exomes and gene panels, as well as the collection of exceedingly larger cohorts.
We are excited that our upcoming product releases will showcase our ability to scale to the demands of those pushing these boundaries.
VarSeq’s V8 Engine Gets a Turbocharger
VarSeq’s strength has always been demonstrated in the immediacy of your data and the complete customization you have over your annotation, filtering and reporting workflow.
From the beginning, the early adopters of VarSeq ranged from clinical labs working with high throughput targeted gene panels to specialized researchers digging into complex families on exomes and genomes.
As VarSeq matured into a trusted solution by many labs and institutions, it has also been expected to handle larger and larger data sets.
Over the past year, we have been putting considerable R&D resources into optimizing, removing limitations and scaling VarSeq core architecture to handle these larger datasets.
A cornerstone of this work is the completely rewritten scalable table featured in our next VarSeq release, but many other areas from import to export have also been finely tuned.
VarSeq can now scale to analyzing whole genomes and thousands of samples.
dbSNP is the most comprehensive catalog of observed mutations in human genetics with over 150 million records. You wouldn’t expect you can work on this size dataset like you would a single NGS samples on your own computer.
With our new version, we can import dbSNP and compare the differences between versions, perform filters and run exports. Our data curation team does this sort of work to vet every new release of public data sources, and by doing this careful version to version comparison has found and reported data issues to NCBI and others (they are good about resolving these issues).
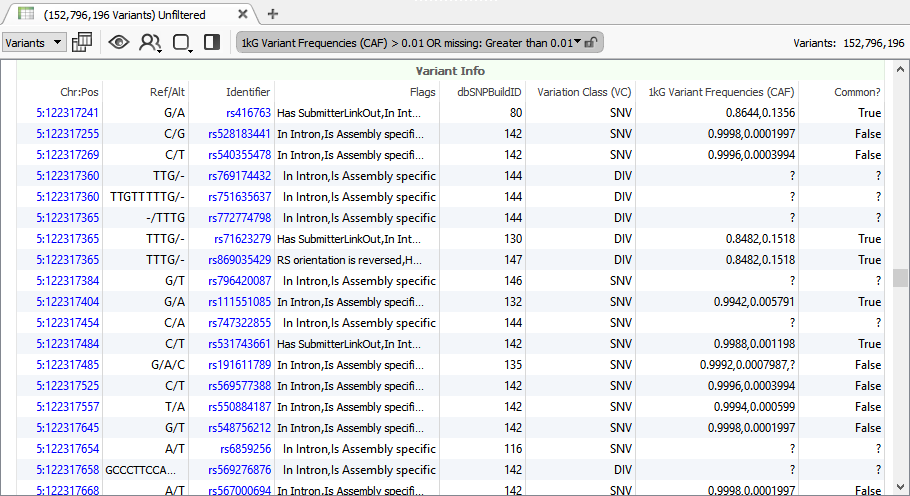 Browsing 150 million variants has never been so easy!
Browsing 150 million variants has never been so easy!
VarSeq has also grown a number of useful tools to summarize information about large numbers of imported samples.
Our Allelic Counts algorithm provides allele and genotypes counts of your imported samples, optionally broken down by sample groups like Affected vs Unaffected.
Our upcoming VarSeq releases adds to that new cohort level Computed Fields, allowing custom summary formulas to be computed across sample numeric fields. For example, you can compute per-variant means and standard deviations of things like strand-bias and read depth to create a custom annotation track useful for referencing and flagging outliers in your new sample workflows.
VSWarehouse Stands Out Because It Stands Alone
VSWarehouse was designed from the ground up to scale to the demands of labs that never want to throw away the results of their NGS sequencing pipeline.
With our upcoming VSWarehouse release, we are excited to have major speed improvements to the initial import and sample-adding routines. With a large number of samples, speedups of 2-10x can be expected.
Labs across the world have been choosing VSWarehouse after carefully considering their alternatives and have been impressed with its capabilities.
VSWarehouse has a number of properties completely absent in an equivalently scalable solution from an enterprise data warehouse:
- Versioning of projects. Adding new samples creates a new version, complete with a snapshot of the relevant annotations and new allelic counts. Previous versions remain accessible as reference points and historical context to clinical decisions.
- Efficient data storage. Using our domain optimized columnar and compressed data format, these full data sets require very modest disk requirements and their self-contained nature make it easy to add to a data backup and archival strategy.
- SQL access of single-table data model. Solutions that try to provide an SQL interface to a variant warehouse often throw every genomic annotation source into a separate table, making basic queries a complex mess of dozens of table joins. Our VSWareouse SQL interface makes a project appear as a single queryable SQL table, while efficiently executing the query directly on the columnar tables underneath the hood.
- Domain specific high-speed import and export utilities. Not only are our multi-core enabled imports and exports extremely fast, they contain a wealth of genomic and bioinformatics expertise in how they normalize variants, merge samples and auto-compute useful fields. Any solution without these properties leaves you working outside the system to prepare your genomic data for import.
Researchers Love SVS Because SVS Keeps Up with Them
Last, but certainly not least, I want to mention scaling in the context of our research focused genomic association and population studies product SNP & Variation Suite, more widely known as SVS.
SVS has always been very memory efficient, but algorithms can be inherently memory intensive.
This has been the case with the matrix operations that are NxN in their dimensions. When the number of samples (N) in certain fields such as agrigenomics have grown to many tens of thousands, it would appear that such algorithms would require computers with hundreds of gigabytes of RAM to run.
It turns out, with the application of advanced mathematics, it is possible to break down these large matrices into smaller ones and still perform the same operations.
I recently wrote about how we did just that in our latest version of SVS to make Kinship Matrices and GBLUP computations feasible on large cohorts on reasonably priced hardware.
Seeing Is Believing, So Come and See
In the upcoming webcast next week, I will expand on these themes while showcasing the latest versions of VarSeq, VSWarehouse and SVS that are scaling to the demands of modern genomics.
Please join us by registering here!