In continuation of our blog posts focusing on new features of VarSeq v2.2.2, here we will discuss the Latest Sample Assessment algorithm for both single nucleotide variants (SNVs) and copy number variants (CNVS). This algorithm annotates the variants of the project with the latest assessment from your variant catalog, which will show the history of interpretations made for the variants as an annotation source in the variant table.
This algorithm can be accessed by going to Add>Computed Data>Latest Sample Assessment algorithm for either Variants or CNVs (Figure 1). Both of these options can be selected by changing the Table Type: drop-down menu in the computed data dialog.
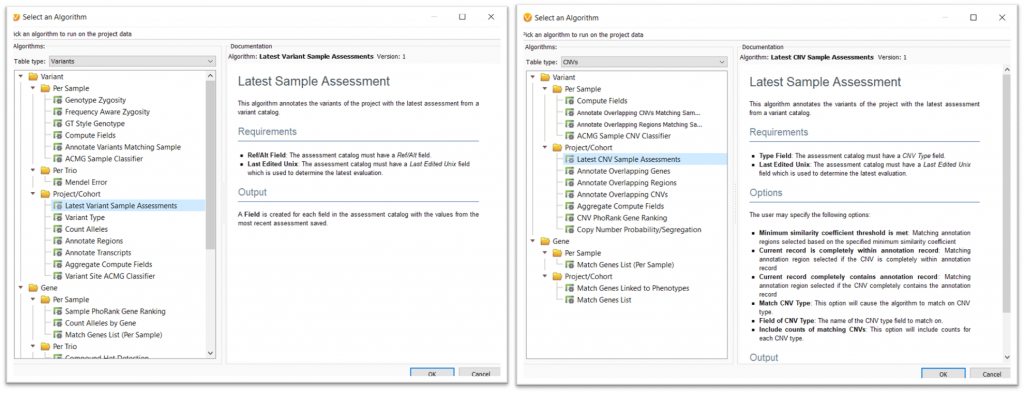
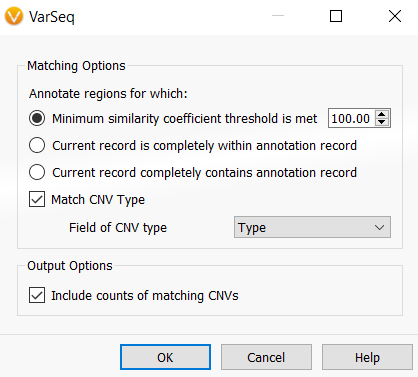
Once you click OK, you can then select the assessment catalog for the algorithm to use. If you are planning to use this algorithm for CNVs, there will then be an additional Matching Options interface, as seen in Figure 2. This dialog allows the user to specify how or when a CNV will be annotated and output how many times a CNV has been observed.
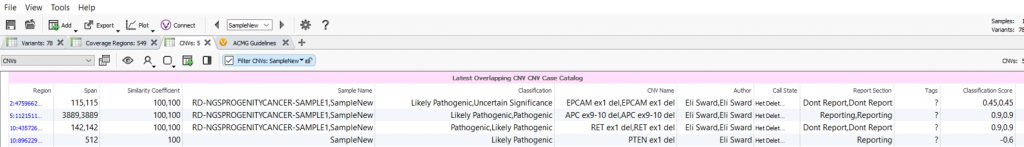
Once the options are selected, this algorithm will appear as an annotation source in your variant or CNV table. In this example, the algorithm was performed on the CNVs in the project and a field was created for each field in the assessment catalog with the values for the most recent assessment saved (Figure 3). As shown in Figure 3, there are multiple interpretations made for the called CNVs as well as the author who created the evaluation.
Together, this algorithm displays the collective repository of changes to variants stored in your assessment catalog and can serve as a history log of all variant interpretations. Hopefully, this feature will provide additional insight into your variant analysis and improve your interpretations of next-generation sequencing variants. As always, if you have any questions about this algorithm or other features of our software, please don’t hesitate to reach out to support@goldenhelix.com. Feel free to also check out some of our other blogs that always contain important news and updates for the next-gen sequencing community.