One of the inherent realities of next-generation sequencing is the ongoing updates to the human reference genome—one of the strongest recommendations to take the original sequencing data and remap to the latest genome assembly. However, there are several reasons why remapping may be impractical. So, an alternative solution is needed to convert the data running through an initial mapping to an earlier assembly (GRCh37, for example) and make it concordant with new genomic coordinates for the latest assembly (GRCh38). Fortunately, Golden Helix has implemented the necessary Liftover functionality to ease this conversion process. There are a few instances where the conversion can be applied, whether you are creating a new project, converting custom databases/annotations, or automated project creation via VSPipeline. One thing must be clearly stated; however, the application of liftover is specific to the VCF based data and associated coordinates for the variants. Unfortunately, liftover will not be able to handle the remapping of reads in the BAM file. Realigning the BAM reads would require a rerun in the secondary pipeline against the current genome assembly. Let’s now explore the liftover options when creating a new project in VarSeq.
VarSeq Project Creation – GRCh37 liftover to GRCh38
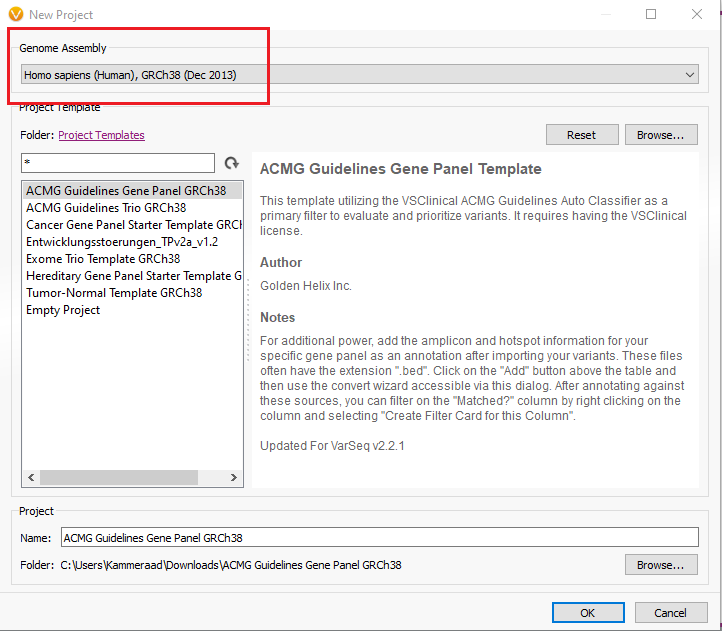
One major goal with VarSeq is to create a workflow template that can be deployed for all incoming samples over time, so the user isn’t left to create the workflow manually with each new project. Regarding liftover, it is important to point out that the template is locked down for the choice assembly. In other words, users will want to create a template specifically dedicated to handling the conversion of GRCh37 based variants to GRCh38. Figure 1 shows the location of the genome assembly selector when creating the project, which is set to the desired final assembly GRCh38. Figure 2 then shows where the liftover tool is located in the import process.
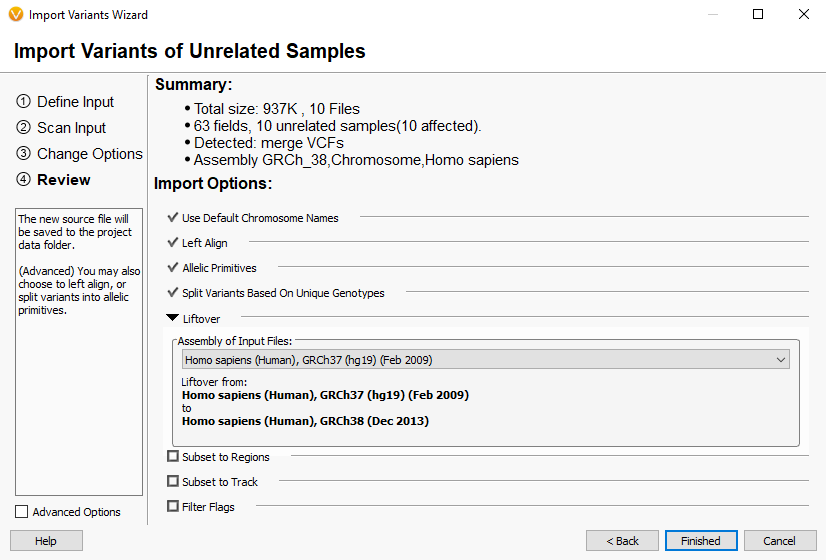
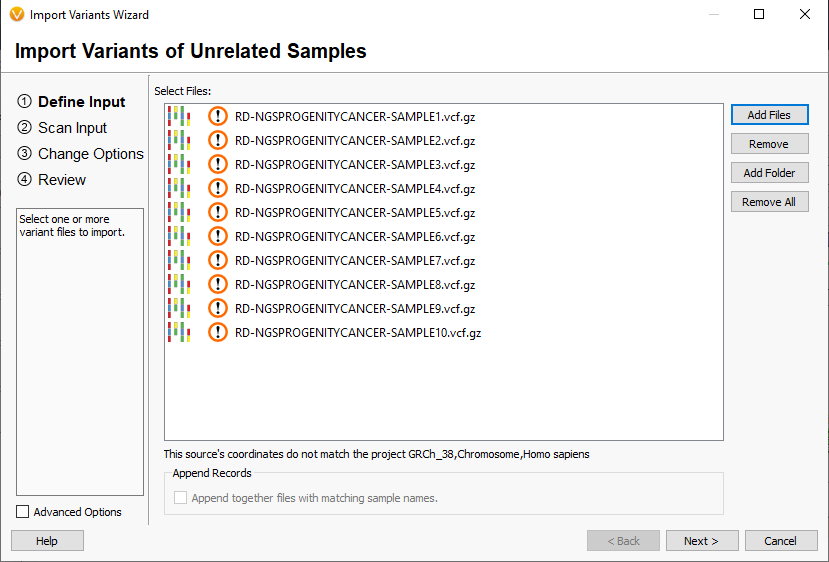
A nice feature recently added in figure 3 is the obvious warning that there is a genome assembly and VCF data mismatch if the user accidentally selects the incorrect assembly.
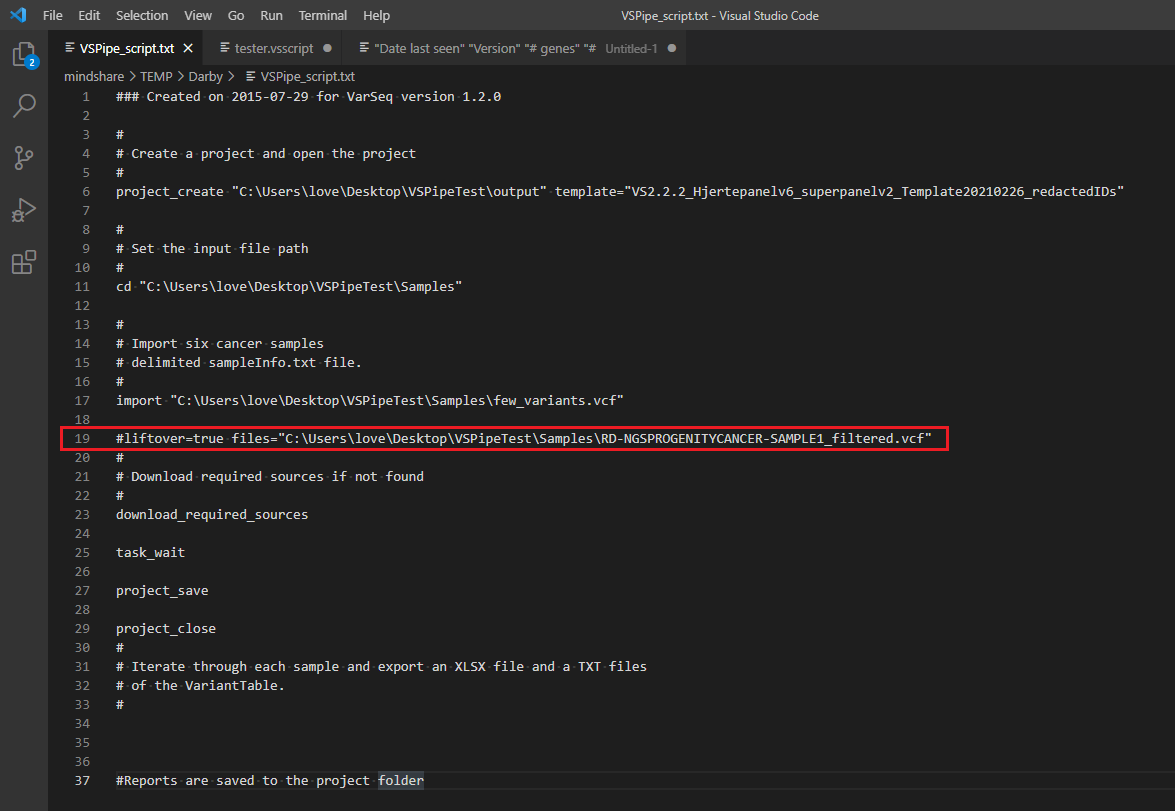
Additionally, we have recently added the liftover option in the batch run process for our users who have automated their clinical workflows via VSpipeline. You’ll see this function added on line 19 of this example batch script seen in Figure 4.
Converting Custom Annotations with Liftover
One convenient tool in VarSeq is adding custom annotations to our list of publicly curated databases using the convert wizard. This spans many types of annotations, region tracks (bed files), custom gene lists, and general variant-based sources. The liftover function has also been implemented in the convert wizard as well so your custom tracks can be updated to the latest assembly. You’ll find this option in the last window of the conversion process, seen in Figure 5.
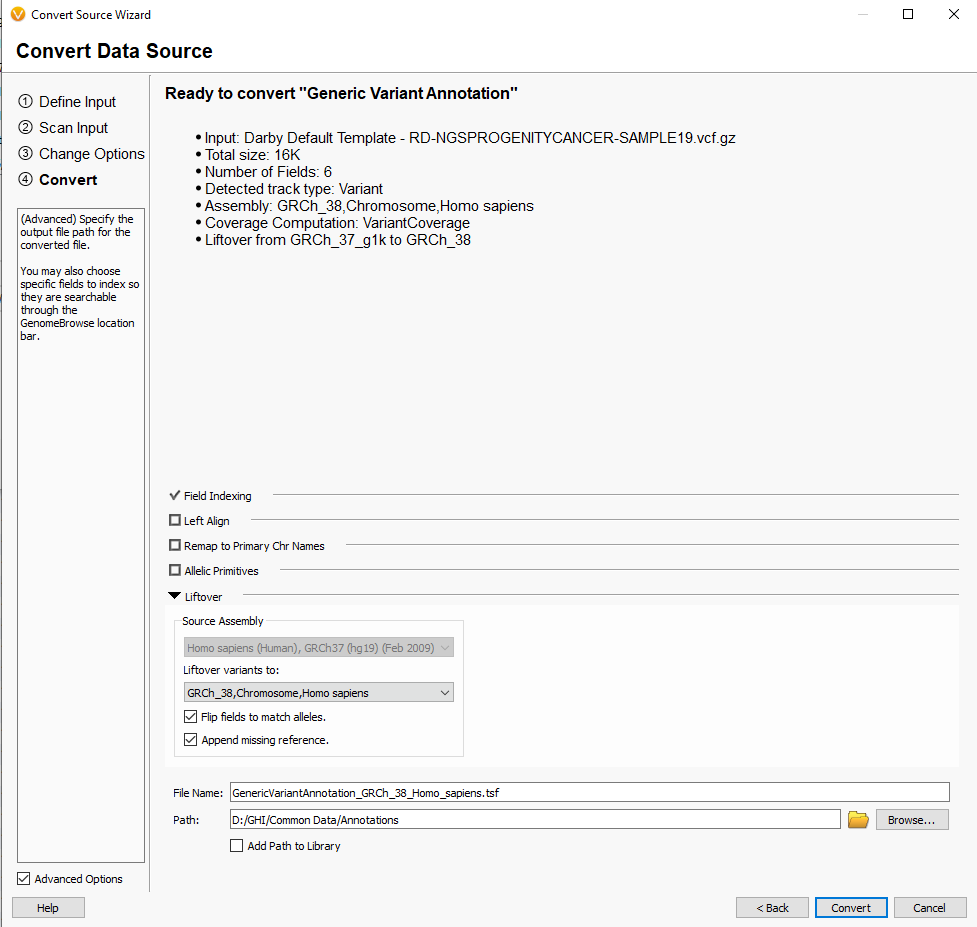
Please explore this new liftover tool if you seek to update your GRCh37 based data into the GRCh38 coordinates. Also, please feel free to contact us at support@goldenhelix.com if you would like formal training on the process. If you enjoyed this content, please check out some of our other blog posts, which contain important information and updates on our clinical interpretation capabilities. Thank you for reading this blog post, and we look forward to hearing from you.