
Next-Generation Sequencing (NGS) technology has decreased the price of nucleotide sequencing exponentially in the last 10 years. The clinical applications are broad, from diagnosis of rare diseases, to carrier screening and hereditary disease risk, and finally, for the personalized treatment of cancer with molecular profiling of tumors for therapeutic, diagnostic, and prognostic genomic biomarkers.
Discovery and Sharing of the Reference Genome for SARS-CoV-2
With sequencing machines broadly available to clinical and research labs, the identification and sequencing of the complete genome of novel pathogens can be done by small groups in a matter of days. Shortly after the outbreak of severe illness, Chinese scientists were able to identify a novel coronavirus in samples taken from the first patients in Wuhan province. Using standard NGS machines and a metagenomic RNA sequencing protocol, a complete genome of the novel virus was assembled and shared with the world on January 12, 2020, by the Chinese authorities. A second group corroborated this finding shortly thereafter. On January 29, 2020, five days after the French Ministry of Health confirmed the first cases of the Wuhan coronavirus, the Institute Pasteur sequenced and shared the whole genome of the coronavirus of two of the first three confirmed cases in France (2019-NCOV Press Release). In comparison, in 2004 it was considered a heroic feat of the international consortium of scientists to sequence the SARS virus genome in 31 days after the investigation of the virus outbreak started (WIRED, 2004).
The final genome of sequenced SARS-CoV-2 consists of a single, positive-stranded RNA that is 29,811 nucleotides long, broken down as follows (GenBank Accession MG772933, RefSeq Accession NC_045512):
- 8,903 (29.86%) adenosines
- 5,482 (18.39%) cytosines
- 5,852 (19.63%) guanines
- 9,574 (32.12%) thymines
Golden Helix’s Data Curation Team brings together genome assemblies and relevant annotations of many species beyond human. Over the course of many years providing support for research and clinical genome analysis applications, it has published curations of over 100 genomes and 1,700 unique annotations including gene models, functional evidence, variation frequencies and clinical interpretations. The SARS-CoV-2 genome and gene model was curated and published within 24 hours of the first customer request.
Other genomes in the infectious disease space have previously been curated by Golden Helix. In figure 6, you find a few examples.
| COMMON NAME | GENOME SIZE |
| Malaria parasite | 23Mbp |
| Staphylococcus aureus | 2.9MBp |
| Tuberculosis causative agent | 4.4Mbp |
| SARS-CoV-2 | 29.9Kbp |
| Ebola virus | 18kbp |
Fig 6: Examples of curated pathogen reference genomes
A Golden Helix user interested in doing analysis on a novel genome has a couple of options. They can contact our Support Team and expect a short turn-around for the curated and published genome to be available for global access. Alternatively, there are existing capabilities built into the software to curate new reference sequences with a guided user-friendly wizard.
Visualization of Reference and Gene Model
Golden Helix provides different analysis tools for research and clinical genomic analysis applications. For researchers, SNP & Variation Suite (SVS) provides general purpose data normalization and annotations, statistical analysis for a broad set of research questions and expansive visualizations for exploring analysis results.
On the clinical side, VarSeq and VSClinical support the analysis of individual patients’ genomic variants for the entire spectrum of clinical genomic tests ranging from focused gene panels, exomes, to complete genomes for hereditary and cancer workflows. Both of these tools embed an integrated genomic visualization tool, GenomeBrowse that is also available as standalone application, free for researchers. Figure 7, below, demonstrates the visualization capabilities of GenomeBrowse for the curated reference genome and genes for SARS-CoV-2 (see fig 7).
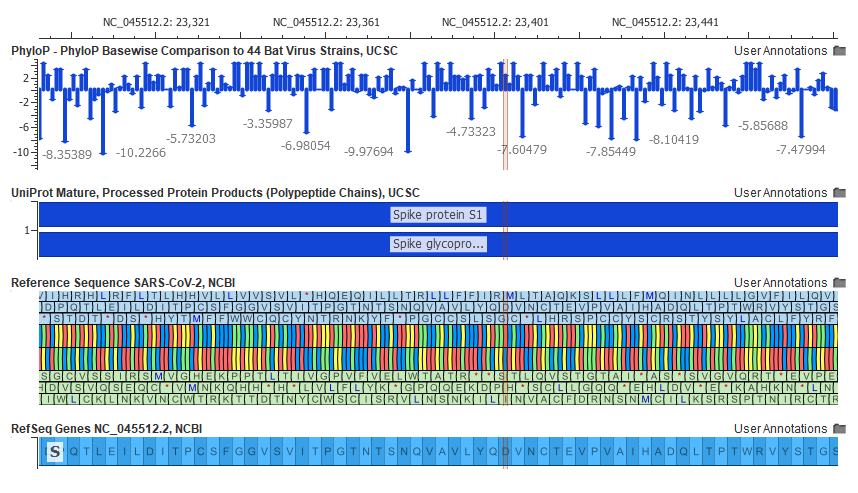
Fig 7: Reference Sequence and Gene Model of SARS-CoV-2 Viral Genome, zoomed to conserved region of Spike Protein S1 with GenomeBrowse
NGS Analysis Pipeline
Now that a reference sequence has been established, easier comparisons can be made between samples and standard analysis workflows and tools for processing NGS data can be employed. This processing, often called “secondary analysis,” takes the millions of short (often 150 nucleotides) sequences produced from the machine and “align” or map them to their position on the reference genome. When multiple reads overlap, the confidence in the true sequence of the sample is raised at that position. Sometimes, individual letters in a sample will be different than the reference, causing a “variation.” A tool specializing in modeling the chance that these variations are real and not technical artifacts of one form or another is called a variant caller, and it produces a Variant Call File (VCF) that can be processed in conjunction with the variants of other samples in a downstream analysis tool such as SVS or VarSeq.
Golden Helix provides industry-leading secondary analysis tools for doing the following industry-standard workflow for SARS-CoV-2 NGS samples:
- Align reads to the reference genome using the BMA-MEM short read alignment algorithm (Li, 2013)
- Remove unwanted identical (duplicate) reads caused by PCR amplification
- Call variants using an accelerated GATK algorithm implemented by Sentieon (Poplin et al. 2017)
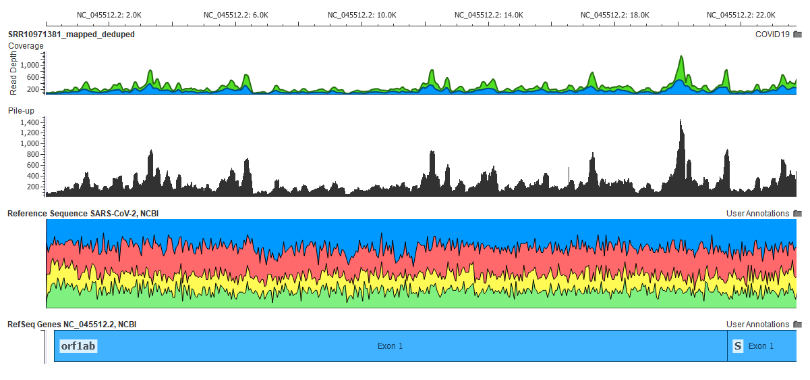
Fig 8: The Wuhan patient’s meta-transcriptomic NGS reads aligned to the SARS-CoV-2 Genome created from this sample
Here is what the alignment of the raw NGS data taken from the first Wuhan patient’s meta-transcriptome looks like aligned to the SARS-CoV-2 genome following this workflow (see Fig 8).
If you enjoyed this preview of our new eBook and wish to continue reading, download a complimentary copy on our site: