When using VarSeq; annotations, application settings, and assessment catalogs are all stored locally. Sometimes these resources can grow to large space grabbing directories, causing you to either purchase additional storage devices or getting rid of previously downloaded resources you might need down the road. But there’s hope! You can set where you want all of your data stored to be… Read more »
We find ourselves talking about our partnership with Sentieon, a lot! More specifically, we are always extolling the powerful, comprehensive genomic data analysis solution we are able to offer our clients. Sentieon’s Secondary Analysis Suite made a significant improvement in runtime over BWA-MEM, GATK, Mutect, and MuTect2 while providing deterministic and identical results. Here are two fantastic white papers that… Read more »
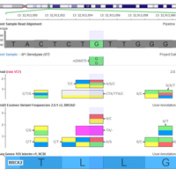
The VarSeq clinical platform is built on a strong foundation of data curation and annotation algorithms to ensure the variants identified have all the information required to make the correct clinical assessments. It’s easy to make light of “variant annotation”, but the details run very deep into the roots of how we represent genomic data, how public data is aggregated, stored… Read more »
Annotating with gnomAD: Frequencies from 123,136 Exomes and 15,496 Genomes When the Broad Institute team lead by Dan MacArthur announced at ASHG 2016 that the successor to the popular ExAC project (frequencies of 61,486 exomes) was live at http://gnomad.broadinstitute.org/, I thought their servers would have a melt-down as everyone immediately jumped on and started looking up their favorite genes and… Read more »
It may have been easy to miss in the drum-beat of monthly annotation updates we do here at Golden Helix, but there are a couple of things that are very special about the January update to the ClinVar database: We added new fields including HGVS names of variants and citations in PubMed for variants ClinVar nearly doubled in size by… Read more »
Variant interpretation is an integral part of any workflow that results in some decisions being made about the validity and suspected functional impact of a variant in a given sample and their presenting phenotypes. The VarSeq Assessment Catalog functionality is designed to assist the VarSeq user in streamlining this process. To include this functionality in your workflow, you will first… Read more »
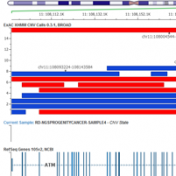
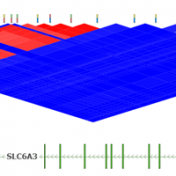
ExAC CNVs were released publicly with a recent publication, providing the full set of rare CNVs called on ~60K human exomes. While there are many public CNV databases out there, this is the first one that was derived from exome data, and thus includes both extremely rare and very small CNV events. With the recent release of Golden Helix’s CNV calling… Read more »
While clinical assessments of germline mutations have been collected in ClinVar under the stewardship of the NCBI and the collaborate effort of many testing labs, the same type of resource has been missing for mutations that could informal clinical care in Cancer. Or at least, that is what I thought until I started to work with CIViC. With the stewardship of… Read more »
One of the tools at the top of the toolbox for researchers working with microarray data is genotype imputation. Genotype imputation is the process of inferring the genotype of one or more markers based on the correlation pattern (aka linkage disequilibrium or LD) of the surrounding markers for which genotypes are known. We have now integrated a natively ported version of BEAGLE into Golden… Read more »
In our SVS 8.6.0 release, we updated our Annotate and Filter Variants feature to utilize our powerful VarSeq annotations. Annotations can be run against gene, interval, variant, and tabular tracks, including RefSeq, ClinVar, CADD, OMIM and OncoMD. The new streamlined dialog allows users to select track specific options and to set up custom filters. While our public annotation repository has… Read more »
Pruning your data based on Linkage Disequilibrium (LD) values is an important quality assurance step for GWAS analysis. In particular, some tests such as Identity by Descent Estimation (IBD), Inbreeding Coefficient Estimation (f) and Principal Component Analysis (PCA) will obtain better results if the markers used are not in linkage disequilibrium with each other. Therefore, Golden Helix’s SVS provides the… Read more »
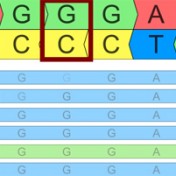
Variant Normalization: Underappreciated Critical Infrastructure It may surprise you to learn that every variant in the human genome has an infinite number of representations! Of course, although true, I’m being a bit hyperbolic to prove a point. Even seemingly simple mutations like single letter substitutions are legitimately represented differently in the local context of other mutations that can be described… Read more »
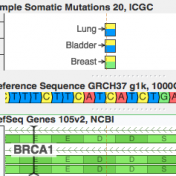
ICGC’s database is now available For quite a while, COSMIC has been synonymous with the catalog of “known somatic mutations”. It is the ClinVar of cancer mutations and invests heavily in “expert curation” (having human experts read papers and pull out references to published somatic mutations). But it turns out there is a new kid on the block, and he… Read more »
Our webcast yesterday featured two clinical workflows and and the ease in moving from an unfiltered variant file to a clinical report containing the variants of interest using VarSeq and VSReports. There were several great questions and I wanted to pass on a few of particular interest. Question: Are annotation sources included in VarSeq for free?
The next release of VarSeq will ship a new product that is highly relevant to our customers in clinical testing labs. Via VSReports, VarSeq now has the ability to generate clinical-grade reports. These reports are fully customizable, containing focused and actionable data. VS Reports ships with report templates that are modeled off of the ACMG guidelines, the de-facto gold standard… Read more »
Have you ever scratched your head when looking up a variant and it seems like the number you have for its position is one off from what it looks like in the file or database? You may be running into the dreaded world of 1-based versus 0-based coordinate representation! If it’s any consolation, I can promise that all the bioinformaticians… Read more »
Since its early release in early 2012, the population frequencies from the GO Exome Sequencing Project (ESP) – from the National Heart, Lung and Blood Institute (NHLBI) have been a staple of the genomic community. With the recent release of ExAC exome variant frequencies, the ESP has been surpassed as the largest cohort of publicly available variant frequencies (by nearly… Read more »
In 1914 the German cytologist Theodor Boveri coined the phrase “Cancer is a disease of the genome”. At this time his ideas were equally revolutionary as they were highly contested. Fast forward. More than hundred years later, Next-Generation Sequencing effectively permits a highly sensitive analysis of cancer cells. It can help us to understand mutations associated with cancer development and… Read more »
It’s come to my attention in recent weeks, through various customer interactions, that many are not aware of the fantastic functionalities that exist in SNP and Variation Suite (SVS) for large-n DNASeq workflows; this includes large cohort analyses with case/control variables. The data you’ll see below is the publically available 1kG Phase 1 v3 Exome sequences from 1,092 individuals with… Read more »
To say the announcement of Dan MacArthur’s group’s release of the Exome Aggregation Consortium (ExAC) data was highly anticipated at ASHG 2014 would be an understatement. Basically, there were two types of talks at ASHG. Those that proceeded the official ExAC release talk and referred to it, and those that followed the talk and referred to it. Why is this… Read more »