Our latest release of the VarSeq software has had a major upgrade with the addition of the new CNV ACMG guidelines! Here are some recent webcasts we’ve given covering the new guideline tool: Family-Based Workflows in VarSeq and VSClinical A User’s Perspective: ACMG Guidelines for CNVs in VSClinical Not only does VarSeq 2.2.2 come with the new guideline tool, but… Read more »
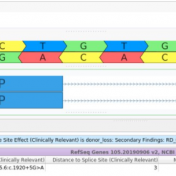
Our latest VarSeq release is one of the largest we’ve ever had, boasting an extensive list of new features and improvements. As part of this release, we have dramatically expanded our support for splice site analysis. This includes improvements to our novel splice site algorithm and support for splice site effect prediction along with several other small improvements. Novel Splice… Read more »
VarSeq 2.2.2 was released on December 17th, 2020 and the main feature that was added to VarSeq was that the VSClinical ACMG Guidelines workflow now has an additional CNV interpretation framework based on the ACMG/ClinGen guidelines. This product supports interpreting CNVs detected with VS-CNV or imported CNVs alongside variants and requires both a VSClinical ACMG license and a CNV license…. Read more »
Those of you who have been attending our recent webcasts have learned about our upcoming VarSeq release. A part of that release will be an additional algorithm that will annotate variants matching the current sample. If you are not familiar with these webcasts, here are several on-demand webcasts I recommend to get you familiar with these new features: Evaluating Copy… Read more »
Golden Helix is excited to release an upcoming VSClinical feature that allows users to analyze next-generation sequencing (NGS) CNV event reporting with ACMG guidelines. This feature will be the first in the NGS workspace to allow this capability and if you are curious about the functionalities you can get a sneak peek by looking at some of our most recent… Read more »
SVS 8.9.0 was released on August 19th and features a new GBLUP by Bin feature and a new utility to find the LD scores of markers and categorize them into bins, along with several mixed-model upgrades and many other upgrades, fixes, and polishes. The two new features LD Score Computation and Binning and Compute GBLUP Using Bins, while they can… Read more »
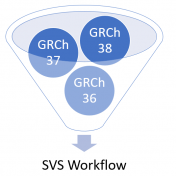
The world has been making a shift to use GRCh38 human genome reference coordinates, but the transition has not been fast. Many of the mainstay human catalog projects are changing to use native GRCh38 catalogs, or are remapping their current data to GRCh38 coordinates. While this seems to be the advancing goal, it is leaving researchers and analysts with the… Read more »
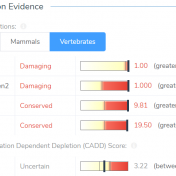
The University of Washington’s Combined Annotation Dependent Depletion (CADD) algorithm measures the deleteriousness of genetic variants. This includes single nucleotide polymorphisms (SNVs) and short insertions and deletions (indels) throughout the human reference genome assembly. This algorithm was introduced in 2014 and has since become one of the most widely used tools to assess human genetic variation. Since 2014, the algorithm has been… Read more »
An under-appreciated area of complexity when looking into the field of genetics from the outside can be found in genes and transcripts. Alternative splicing allows eukaryotic species to have a wonderfully powerful genetic code, resulting in multiple protein isoforms being encoded in a single section of DNA. But when it comes to variant interpretation, different transcripts can result in widely different predicted… Read more »
In trio workflows, one of the most important factors in scoring a variant is understanding how that variant is inherited from the parents. Likewise, when looking at extended families, the segregation, or presence of the variant among the affected versus unaffected individuals provides evidence for its pathogenicity for a given phenotype or disease. Given the nature of Copy Number Variants… Read more »
VarSeq 2.2.1 was released on April 1st and features an upgraded gene annotation capability with new RefSeq genes tracks and an AMP workflow addition: the Drugs and Trials tab. The new RefSeq human genome genes tracks contain updated gene names and the recognition of any MANE (Matched Annotation from NCBI and EMBL-EBI) identified transcripts. VarSeq has been updated to be… Read more »
VarSeq 2.2.0 was released today and this a stable release full of upgrades and polishes. Some of the newer features include the ability to store and include AMP Cancer assessment catalogs on VSWarehouse, quicker accessibility to common annotations plotted in GenomeBrowse, and the addition of all of our standard templates for the GRCh38 genome assembly. Many of the polishes were… Read more »
The SVS 8.8.3 release was created to incorporate some of the CNV, genome assembly control, and splice site capabilities that are present in VarSeq, as well as clean up and streamline the GWAS workflows (like when using Mixed Linear Model algorithms) for a better user experience. New Product Add-Ons for SVS GoldenHelix SVS now includes in-silico splice site, functional prediction… Read more »