Orphanet is a public database available in VarSeq that aims to improve the current understanding and treatment options for patients affected by rare diseases. This resource was established in France in 1997 and has gradually grown to cover a consortium of over 40 countries in Europe. The primary goal of this database is to provide a universal nomenclature to classify… Read more »
Clinical testing labs produce reports as the end product of the NGS variant detection and interpretation workflow. Necessarily, the content, detail, and presentation of the report needs to be specialized to each clinical lab, and potentially each offered test. Our last blog post introduced the new Word-based report templates in VSClinical. In this blog post, we will introduce and explore… Read more »
In many cases, VarSeq users typically run single trio projects or perhaps an extended family project. Not only are all the inheritance model algorithms available in the VarSeq software to capture de novo, dominant, or recessively inherited variants but there are a number of quality control fields to help ensure the pedigree was set up properly. The last thing any… Read more »
Next-generation sequencing generates an immense amount of data which is then subject to a multi-step process to establish a validated bioinformatic pipeline. From processing raw sequence data to the detection of genetic mutations, establishing a validated and consistent bioinformatic pipeline makes a huge difference in the quality of patient care and accuracy of results. In this blog, we are focusing… Read more »
The recent release of VarSeq 2.2.2 brings our Word report template system, previously featured in VSClinical AMP, to the VSClinical ACMG workflow. This blog post will describe how to use the Word template system using one of our shipped templates as well as how to start customizing your own templates. We will cover the three different report templates that ship… Read more »
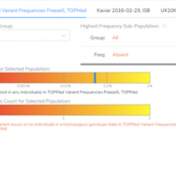
Our previous webcast demonstrated some of the new functionalities of VSClinical, including the ability to add ACMG frequency sources for the ACMG BA1, BS1, and PM2 criteria. This new feature was spurred by the feedback from our users, which requested supporting frequency tracks other than gnomAD Exomes and 1kG Phase3. Now, users can implement population catalogs to VSClinical such as… Read more »
In this blog post, I will be analyzing a loss-of-function splice variant in MTHFR using VarSeq. In the search for clinically relevant variants contributing to rare disorders, efficient filtering strategies are an important step in eliminating disinteresting variants. However, any applied filters must also ensure no interesting variants inadvertently get filtered out. Golden Helix provides the tools to complete this… Read more »
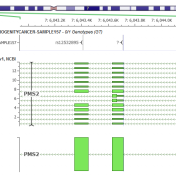
In the webcast, Evaluation of Copy Number Variants with VSClinical’s New ACMG Guideline Workflow, we discussed how VSClinical implements Section 4 of the ACMG guidelines. Specifically, we focused on integrating literature and publications to assess the pathogenicity of a CNV event when there was a lack of dosage sensitivity information. One of the primary pieces of evidence for evaluating genes… Read more »
The potential of genetic testing to impact a patient’s life has been greatly accelerated by the sharing of variant interpretations done by clinical labs in public repositories such as ClinVar. This is not an inevitable outcome, but the persistent work and advocacy of people like Dr. Heidi Rehm and organizations like ClinGen. We recently participated in a survey and vetting… Read more »
As clinical genetic tests have been adopted as a critical enabler of precision medicine, the number of tests offered by clinical labs and the volume of tested patients has grown by orders of magnitude in the past five years. The Gene Testing Registry, managed by the NIH, documented a rise from 13,000 to 60,000 tests offered in the US market… Read more »
Overview VSClinical enables users to evaluate variants according to the ACMG guidelines in a high-throughput fashion and obtain consistent results and accurate variant interpretations. This feature is tightly integrated into our VarSeq platform as well, and when paired together, users can evaluate NGS data and obtain clinical reports all in one suite. Coupled with the ability to find novel or… Read more »
VSClinical provides a rapid-fire way to investigate any variant’s impact by following the ACMG Guidelines process for classification. We will be demonstrating this by looking at interesting examples of rare disorders and showcasing some evaluation steps users may deploy in their analysis. Our first example in this blog series is for a patient who has an indifference to pain, while… Read more »
Functional Predictions and Conservation Scores in VSClinical Several algorithms have been developed to predict the impact of amino acid substitutions on protein function and quantify conservation of nucleotide positions. These methods provide vital supporting evidence to clinicians when interpreting variants in accordance with the ACMG guidelines. The two most popular functional prediction algorithms are SIFT and PolyPhen2, while the most… Read more »
There are many good reasons why the pursuit of the highest quality genomic interpretation would lead you to the latest human reference. It is more complete and fixes incorrect or partially missing genes that have known implications for human disease. While most major projects cataloging human populations have plans to re-do all their genomic alignments to the new human reference… Read more »
Interpretation of variants in accordance with the ACMG guidelines requires that variants near canonical splice boundaries be evaluated for their potential to disrupt gene splicing [1]. The five most common tools for splice site detection are NNSplice, MaxEntScan, GeneSplicer, HumanSplicingFinder, and SpliceSiteFinder-like. Because these algorithms have been made easily accessible in the bioinformatics tool Alamut, they have been canonized for… Read more »
Dr. Suzanne Lewis is a Clinical Professor in the Department of Medical Genetics at University of British Columbia (UBC) and Senior Investigator at British Columbia Children’s Hospital Research Institute (BCCHR), Vancouver, Canada. She is also the Chair of the iTARGET Autism Project and Vice-Chair of Autism Canada Chief Medical Officer and VP Research of Pacific Autism Family Network. She and… Read more »
In case you missed our live event yesterday, I wanted to share the Q&A session and a link to the webcast recording: An Exploration of Clinical Workflows in VarSeq. Question:You mentioned saving projects as templates, will it save GenomeBrowse plots in the project template or do you have to replot the data when opening the software? Answer: Yes, if you save your… Read more »
We have been heads down doing the detailed and careful work to improve our CNV caller algorithm in the past three months since our we launched our Exome capable CNV caller and are very excited about the massive step forward we have made with the VarSeq 1.4.5 release. Additionally, we have added the all new Whole Genome large-event caller capable… Read more »
Annotating with gnomAD: Frequencies from 123,136 Exomes and 15,496 Genomes When the Broad Institute team lead by Dan MacArthur announced at ASHG 2016 that the successor to the popular ExAC project (frequencies of 61,486 exomes) was live at http://gnomad.broadinstitute.org/, I thought their servers would have a melt-down as everyone immediately jumped on and started looking up their favorite genes and… Read more »
It may be possible to say that annotating a variant correctly and accurately against gene transcripts is the most important job of a variant annotation and interpretation tool. We take it very seriously at Golden Helix as we support VarSeq and its use by our customers in both research and clinical contexts. It has been a source of frustration that… Read more »