Variant Normalization: Underappreciated Critical Infrastructure It may surprise you to learn that every variant in the human genome has an infinite number of representations! Of course, although true, I’m being a bit hyperbolic to prove a point. Even seemingly simple mutations like single letter substitutions are legitimately represented differently in the local context of other mutations that can be described… Read more »
When the new human reference genome was released over two years ago, it was hailed as a significant step forward for next generation sequencing. Compared to GRCh37, the new GRCH38 reference assembly fixed gaps, repaired incorrect sequences and offered access to sections of the genome that had been previously unaccounted for. Despite these improvements, adoption of the new assembly has… Read more »
There used to be much energy expended at conferences, bioinformatics forums and even publications about what was the better strategy for interpreting variants of clinical significance: Rule-based filtering and classification mechanisms or rank-based prioritization through all-encompassing “pathogenicity” scores. Both have shown to be effective. Rule-based systems, as exemplified in this filtering diagram in Baylor’s ground-breaking paper on clinical whole-exome sequencing… Read more »
Solving the Eigenvalue Decomposition Problem for Large Sample Sizes Since our introduction of the mixed model methods in SVS, along with GBLUP, we have been very pleased to see it used by a number of customers working with human and agri-genomic data. As these customers have grown their genomics programs, the number of samples they have for a given analysis… Read more »
Submit directly to N-of-One from VarSeq If you or your lab uses N-of-One solutions for clinical annotations, here’s some good news: You can now submit directly to N-of-One from VarSeq! N-of-One’s set of preferred transcripts may differ from those outputted by our algorithms in VarSeq, so our solution was built with that in mind. Our slick, easy to use, and… Read more »
Concepts and Relevance of Genome-Wide Association Studies Genome-Wide Association Studies continue to be a very effective method for determining the underlying cause of disease, Golden Helix is happy to share our long-standing knowledge with the community. We are very happy to announce that “Concepts and Relevance of Genome-Wide Association Studies”, a paper surrounding GWAS was recently published in Science Progress… Read more »
As VarSeq continues its adoption amongst clinical labs and researchers looking for reproducible workflows for variant annotation, filtering and interpretation, we have continued to prioritize the addition of features to assess the quality of the upstream data at a variant, coverage and now sample level. The Importance of Quality Assurance Sample prep and sequencing problems are difficult to detect through the analysis… Read more »
Genetic Data Warehousing e-Book In our webcast last week, we announced the upcoming release of VSWarehouse, a scalable genetic data warehouse for VarSeq. Genetic data warehousing becomes more important as Next-Generation Sequencing is taking off in the clinic, creating significant data management issues for clinicians, scientists and IT professionals alike. How can we retain the massive amounts of data coming out… Read more »
Compound Heterozygous Workflows: Including a 2nd Affected Child Looking for Compound Heterozygous regions for a trio is fairly straight forward in VarSeq, we include this workflow in our shipped Exome Trio Template. An example of which is included with our Example Projects which can be found by going to File > Example Projects > Example YRI Exome Trio Analysis. But… Read more »
I hope your 2016 is off to a good start! We at Golden Helix have been busy preparing and planning new and updated content for the community. First up is an updated GWAS ebook which includes a sample Meta-analysis example project. In the 2nd edition, we start with an introduction to GWAS exploring its biology and origins as well as… Read more »
The most common use of the VarSeq Match Gene List algorithm of course is to determine if the variants in your data set are contained within your genes of interest. As an example of this, say you are working with a whole exome trio and only want to consider those variants that are contained within the 56 genes recommended by… Read more »
As VarSeq’s adoption has grown among analysts using whole exome data to diagnose rare diseases, a couple of family designs outside of the common trio of an affected child and both parents have come up frequently. While having both parents provides the maximum power to discover de novo mutations and recessively inherited variants, it is not always possible to contact… Read more »
With the release of VarSeq 1.3.1 we have included a new demo project to showcase a single tumor-normal pair analysis workflow. The project can be accessed through VarSeq and VarSeq Viewer by going to File > Open Example Projects > Example Tumor-Normal Pair Analysis. This project contains an exome pair (Normal-N990005 and Tumor-T990005) from the Gastric Cancer study Exome sequencing of… Read more »
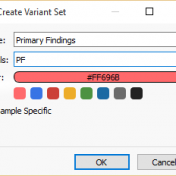
With the release of VSReports, we added the ability to “select” rows of your filtered output (often variants, but potentially things like coverage regions or genes) with a new feature dubbed “Record Sets”, but more often described as “colored checkboxes” for your tables. Although necessary for the important task of marking primary, secondary or other sets of variants for a… Read more »
As VarSeq gains in popularity, we want to give Viewers and customers alike the opportunity to look at projects that are completed from start to finish. To this end, VarSeq (and VarSeq Viewer!) currently comes with two demonstration projects, Example TruSight Cardio Gene Panel and Example YRI Exome Trio Analysis. To access these projects from the VarSeq start page go to… Read more »
The next release of VarSeq will ship a new product that is highly relevant to our customers in clinical testing labs. Via VSReports, VarSeq now has the ability to generate clinical-grade reports. These reports are fully customizable, containing focused and actionable data. VS Reports ships with report templates that are modeled off of the ACMG guidelines, the de-facto gold standard… Read more »
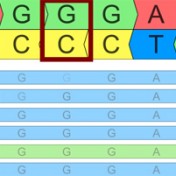
Have you ever scratched your head when looking up a variant and it seems like the number you have for its position is one off from what it looks like in the file or database? You may be running into the dreaded world of 1-based versus 0-based coordinate representation! If it’s any consolation, I can promise that all the bioinformaticians… Read more »
This last week I had the pleasure of attending the fourth annual Clinical Genome Conference (TCGC) in Japantown, San Francisco and kicking off the conference by teaching a short course on Personal Genomics Variant Analysis and Interpretation. Some highlights of the conference from my perspective: Talking about clinical genomics is no longer a wonder-fest of individual case studies, but a… Read more »
There are many approaches that one might use to define a variant as potentially deleterious. For example, we often see analysis workflows based on rare, non-synonymous variants, perhaps incorporating additional annotation sources that capture known or predicted consequences of coding variants. Annotations for coding regions of the genome are relatively abundant and familiar to genome scientists. We are comfortable in… Read more »
There’s a strong desire in the genetics community for a set of canonical transcripts. It’s a completely understandable and reasonable thing to want since it would simplify many aspects of analysis and especially the downstream communicating and reporting of variants. Unfortunately, biology isn’t so tidy as to provide a clear answer for which transcript is the important one. Consequently, there… Read more »