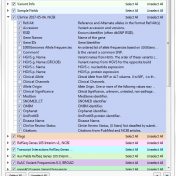
Revisiting the Five Splice Site Algorithms used in Clinical Genetics Interpretation of variants in accordance with the ACMG guidelines requires that variants near canonical splice boundaries be evaluated for their potential to disrupt gene splicing [1]. The five most common tools for splice site detection are NNSplice, MaxEntScan, GeneSplicer, HumanSplicingFinder, and SpliceSiteFinder-like. Because these algorithms have been made easily accessible… Read more »
2017 was an incredibly prosperous year for Golden Helix; we released a handful of new features, announced new partnerships and completed our end-to-end architecture for clinical testing labs. Our webcast series has become a very popular way for our community to stay up-to-date with our new capabilities and best practices in genetic analysis using our software. We had three webcast… Read more »
Clinical Assessment Tracks Golden Helix provides a large catalog of annotation sources for our research and clinical clientele. Making these public data repositories available to all our users is no easy task. As Cody Sarrazin mentioned in his blog post, annotation curation is a complex data science pipeline. This process aggregates data from many disparate sources and normalizes it into… Read more »
Next-Gen Sequencing promised to be the ultimate paradigm when it comes to genetic research and clinical testing since it contains the complete genetic information. When it comes to the current reality in testing labs, there are still a number of additional testing paradigms used in an analysis, specifically, copy number variations. Among these, labs still widely use Chromosomal Microarrays and… Read more »
An Example of an Integrated Clinical Workflow for CNVs and SNVs In this blog series, I discuss the architecture of a state of the art secondary pipeline that is able to detect single nucleotide variations (SNVs) and copy number variations (CNVs) in one test leveraging next-gen sequencing. In Part I, we reviewed genetic variation in humans and looked at the key… Read more »
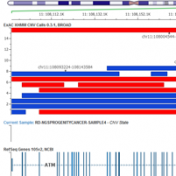
Examples of CNV Calling What do CNV calls actually look like? What are some of the key metrics to determine an event? Part IV of the Secondary Analysis 2.0 blog series will answer these questions by walking through some examples of how our CNV caller, VS-CNV, identifies CNVs. Golden Helix integrates multiple metrics to determine if a CNV event is… Read more »
Detection of CNVs in NGS Data Our Secondary Analysis 2.0 blog series continues with Part III: Detection of CNVs in NGS Data. We will give you an overview of some design principles of a CNV analytics framework for next-gen sequencing data. There are a number of different approaches to CNV detection. The published algorithms share common strategies to solve the… Read more »
In this blog series, I will discuss the architecture of a state of the art secondary pipeline that is able to detect single nucleotide variations (SNV) and copy number variations (CNV) in one test leveraging next-gen sequencing. In Part I, we reviewed genetic variation in humans in general and looked at the key components of a systems architecture supporting this… Read more »
Human genetic variation makes us unique. On average, humans are to 99.9% similar to each other. Understanding in detail what the nature of the difference in our genetic make-up is all about allows us to assess health risks, and eventually enables Precision Medicine as we determine treatment choices. Furthermore, it enables scientists to better understand ancient human migrations. It gives… Read more »
In the past couple of weeks, the topic of the Filter and Quality fields in the popular ExAC population catalog has come up a number of times. It turns out that unlike the 1000 Genomes project, which decided to very heavily filter their variant list to only contain variants they consider high quality, ExAC chose to include more dubious variants… Read more »
Since we released our Phenotype Gene Ranking algorithm in VarSeq, it has become a staple of the way people conduct their analysis. It allows for a combination of filtering with ranking to prioritize follow-up interpretations of analysis results. Our PhoRank algorithm will be available in our upcoming SVS release to also aid in the numerous research workflows performed on SNPs… Read more »
Genome-wide association study (GWAS) technology has been a primary method for identifying the genes responsible for diseases and other traits for the past ten years. GWAS continues to be highly relevant as a scientific method. Over 2000 human GWAS reports now appear in scientific journals. In fact, we see its adoption increasing beyond the human-centric research into the world of… Read more »
ExAC CNVs were released publicly with a recent publication, providing the full set of rare CNVs called on ~60K human exomes. While there are many public CNV databases out there, this is the first one that was derived from exome data, and thus includes both extremely rare and very small CNV events. With the recent release of Golden Helix’s CNV calling… Read more »
December’s webcast will provide the Golden Helix community with a more in-depth look at CNV analysis in VarSeq. On December 7th, Dr. Nathan Fortier will discuss the challenges and metrics surrounding CNV detection and then demonstrate VarSeq’s new capability from VCF to clinical report. Wednesday, December 7th @ 12:00 PM, EST Numerous studies have documented the role of Copy Number Variations (CNVs)… Read more »
One of the tools at the top of the toolbox for researchers working with microarray data is genotype imputation. Genotype imputation is the process of inferring the genotype of one or more markers based on the correlation pattern (aka linkage disequilibrium or LD) of the surrounding markers for which genotypes are known. We have now integrated a natively ported version of BEAGLE into Golden… Read more »
Dr. Sergey Kornilov, a Duncan Scholar in Molecular and Human Genetics at Baylor College of Medicine, combines his broad psychology background with genetics to research the genetic basis of neurodevelopmental disorders with a unique dual perspective. Neuro-developmental disorders, for example, those of the spoken and written language, affect many worldwide – up to 10% of preschool children. In most cases, these… Read more »
Copy Number Variants have been important to clinical genetics for quite a while now. So, what has made now the right time to be looking at calling CNVs from NGS data? Well, there are a number of good reasons. The dominant one is simply that the NGS data you are already creating for calling variants can be used in many cases… Read more »
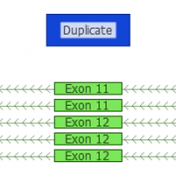
Question: Now that I’ve added annotation sources for my sample, filtered down to a list of interesting variants, flagged those variants and generated a clinical report, can I save or copy the annotation sources and filters for use on another sample? Short Answer: Yes! Long Answer: VarSeq was created with ease and efficiency in mind. In VarSeq, once you’ve defined… Read more »
Now available in SVS! Increasingly important in the analysis of the genotype to phenotype relationship is accurately accounting for the relatedness of samples. This is especially important to model correctly in plant and animal populations where man-directed breeding shapes the relationship structure. Along with trait association, one of the high-value use cases for genotyping animals and plants is to estimate… Read more »
Every month hundreds of clinicians and researchers access the variety of free resources on the Golden Helix website. Our resource library hosts eBooks, webcasts and tutorials to keep the community apprised of new methods, informed on best practices and to help our customers get the most out of their software purchase. Here is a list of the 5 most watched webcasts… Read more »