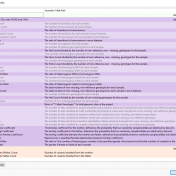
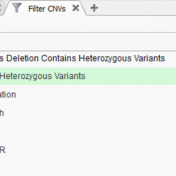
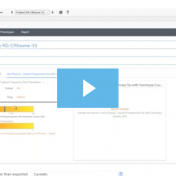
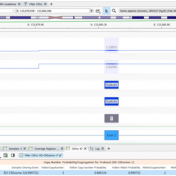
In many cases, VarSeq users typically run single trio projects or perhaps an extended family project. Not only are all the inheritance model algorithms available in the VarSeq software to capture de novo, dominant, or recessively inherited variants but there are a number of quality control fields to help ensure the pedigree was set up properly. The last thing any… Read more »
Our latest release of the VarSeq software has had a major upgrade with the addition of the new CNV ACMG guidelines! Here are some recent webcasts we’ve given covering the new guideline tool: Family-Based Workflows in VarSeq and VSClinical A User’s Perspective: ACMG Guidelines for CNVs in VSClinical Not only does VarSeq 2.2.2 come with the new guideline tool, but… Read more »
Thank you for attending the webinar focused on implementing VarSeq and VSClinical for family-based workflows. If you would like to use the webinar as a reference or were not able to attend, you can access it using the following link to view ‘Family-Based Workflows in VarSeq and VSClinical. Here is a brief recap of what we discussed: This webinar demonstrated… Read more »
There is a multitude of interesting new features that have been incorporated into VarSeq 2.2.2. In this blog, I want to continue the discussion of these features and how each can be incorporated into your workflow, and also discuss the application of the Probability Segregation algorithm for copy number variation (CNV) analysis. The Probability Segregation algorithm is a new algorithm… Read more »
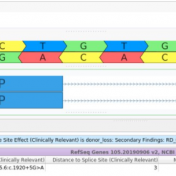
Our latest VarSeq release is one of the largest we’ve ever had, boasting an extensive list of new features and improvements. As part of this release, we have dramatically expanded our support for splice site analysis. This includes improvements to our novel splice site algorithm and support for splice site effect prediction along with several other small improvements. Novel Splice… Read more »
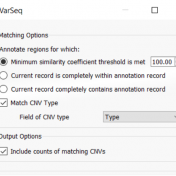
In continuation of our blog posts focusing on new features of VarSeq v2.2.2, here we will discuss the Latest Sample Assessment algorithm for both single nucleotide variants (SNVs) and copy number variants (CNVS). This algorithm annotates the variants of the project with the latest assessment from your variant catalog, which will show the history of interpretations made for the variants… Read more »
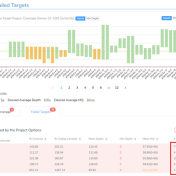
Curious about how coverage statistics can be used in conjunction with VarSeq? Evaluating the coverage over target regions or whole genomes is essential whether you are working with variant or CNV analysis. VarSeq has had the capability to compute sample level coverage statistics for some time now, but in the 2.2.2 release of VarSeq, there are some new features that… Read more »
In this blog post, I will be analyzing a loss-of-function splice variant in MTHFR using VarSeq. In the search for clinically relevant variants contributing to rare disorders, efficient filtering strategies are an important step in eliminating disinteresting variants. However, any applied filters must also ensure no interesting variants inadvertently get filtered out. Golden Helix provides the tools to complete this… Read more »
The potential of genetic testing to impact a patient’s life has been greatly accelerated by the sharing of variant interpretations done by clinical labs in public repositories such as ClinVar. This is not an inevitable outcome, but the persistent work and advocacy of people like Dr. Heidi Rehm and organizations like ClinGen. We recently participated in a survey and vetting… Read more »
As clinical genetic tests have been adopted as a critical enabler of precision medicine, the number of tests offered by clinical labs and the volume of tested patients has grown by orders of magnitude in the past five years. The Gene Testing Registry, managed by the NIH, documented a rise from 13,000 to 60,000 tests offered in the US market… Read more »
Copy Number Variation (CNV) is a type of structural variation in which sections of the genome are duplicated or deleted. Although CNV events are rare in the human population, constituting approximately 10% of the human genome, they are also associated with being causal mutations for disease phenotypes. Because of this, it is important for clinical and research settings to identify… Read more »
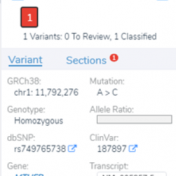
Examples of Clinical Variant Interpretation with VSClinical In this chapter, I’d like to go through a few examples for variants that have been classified with the help of VSClinical. This will give you a better understanding of how data sources are actually being represented in the software and how those are used to make decisions on applicable criteria. It goes… Read more »
Rules for Combining Various Classification Criteria Now that we have a solid understanding of how the various criteria are meant to be applied, it’s time to look at how the evidence collectively leads to the clinical categorization of a variant. Let’s go through the rule framework for combining the various criteria. Pathogenic In order for a variant to be classified… Read more »
Clinical Variant Analysis – Classification Criteria of Benign Variants The classification of benign variants is overall simpler and more straightforward, with the majority of benign variants being eliminated through allele frequency in various population catalogs. BA1 If a variant is common in one or more population catalog, as indicated by the allele frequency associated by the appropriate sub-population, it can… Read more »
Clinical Variant Analysis – Classification Criteria of Pathogenic Variants The ACMG Guidelines are utilized for the interpretation of variants. They are primarily applied to diagnose suspected inherited (primarily Mendelian) disorders in a clinical diagnostic laboratory setting. While evaluating variants no matter what the origin, it is important to distinguish between variants that are pathogenic (i.e., causative) for a disease and a… Read more »
Importance of Quality in Association Tests SVS is a research application platform provided by Golden Helix that enables an array of computational analyses including genome-wide association studies (GWAS). GWAS is an observational study that can provide insight into the association of genetic variants with traits and complex disorders. The foundation of GWAS utilizes large cohorts sequenced with single nucleotide polymorphisms… Read more »
What is Genomic Prediction? Genomic prediction is an algorithm widely used to improve desirable phenotypic traits in agriculture. For example, the cattle industry uses genomic prediction to improve beef quality and palatability as well as improve dairy production (1,2). By using genomic prediction, researchers can minimize multiple expenses in breeding industries as well as diminish the need for performing cumbersome… Read more »
In a recent webcast, our VP of Product and Engineering Gabe Rudy gave us insight into the current capability and benefits to lifting over to the GRCh38 assembly. Golden Helix fully supports this transition into the most recent reference assembly and have developed our tools on both the 38 and 37 fronts. The purpose of this blog is to not… Read more »
VSClinical is our most recent product that allows users to evaluate variants according to the ACMG guidelines. As with any tertiary analysis, there is a need to implement best practices into your workflow and using VSClinical for the ACMG guidelines is no exception. That said, we have put together a Best Practices Blog Series, with the purpose of discussing some… Read more »
Streamlining the ACMG Guidelines and Providing Scoring Recommendations As we discussed in our recent webcast on VSClinical, the process of scoring the ACMG guidelines requires evaluating evidence for the connection between a variant and the disorder or condition being evaluated by the genetic test for an individual. These lines of evidence cover clinical presentation, gene function, bioinformatic annotations and in-silico… Read more »