The University of Washington’s Combined Annotation Dependent Depletion (CADD) algorithm measures the deleteriousness of genetic variants. This includes single nucleotide polymorphisms (SNVs) and short insertions and deletions (indels) throughout the human reference genome assembly. This algorithm was introduced in 2014 and has since become one of the most widely used tools to assess human genetic variation.
Since 2014, the algorithm has been updated 4 times, which brings us to our most recent version, CADD v1.4/v1.5. This new version is integrated into the GoldenHelix VarSeq and SVS software packages, for both GRCh37 (hg19) (CADD v1.4) and GRCh38 (hg38) (CADD v1.5) genome assemblies. We have covered this algorithm in a previous blog so herein we will cover a basic overview of the CADD algorithm as well as unique changes from previous versions and how this is used in our software applications.
CADD Score Background
For any given variant that is aligned to the reference genome assembly, both coding and non-coding, a CADD score is computed using more than 60 different annotation sources. A shown in Figure 1, from (Rentzsch, 2019), these annotations are based on genomic features derived from surrounding sequencing context, gene model annotations, evolutionary constraints, and functional predictions. The raw CADD scores are then computed into PHRED-like rank scores based on the genome-wide distribution for all potential ~9 billion SNVs (3 billion x 3 potential alternative alleles). Thus, there are two CADD scores: Raw and PHRED.

Figure 1: More than 60 annotation sources are used to compute CADD scores
Raw and PHRED CADD Scores
As highlighted in the publication, Raw CADD scores should be used when comparing cases to controls as higher values indicate the SNV is more likely to be deleterious. On the other hand, PHRED CADD scores should be used for causal or fine-mapping variant interpretation within specific loci as they can convey estimated pathogenicity relative to all possible SNVs in the reference genome (Rentzsch, 2019).
RAW and PHRED and Estimated?
In addition to integrating the Raw and PHRED scores, VarSeq also provides an Estimated? column, as shown in Figure 2, which indicates that the CADD score is estimated as the variant is likely a “novel” indel. In other words, an Estimated? value of True indicates that an indel CADD score was not directly available from the approximately 20 million indels already observed in the CADD database derived from 1000 Genomes and is thus estimated. These scores are computed using the scores from flanking or deleted bases surrounding the event and more information on how this is computed can be found in our documentation.
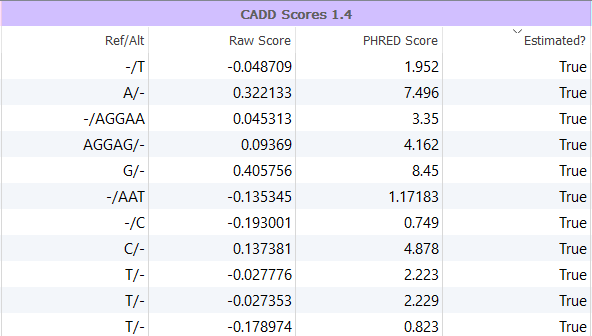
Figure 2: VarSeq integrates precomputed values from the CADD database but also incorporates estimated scores for novel indels
What’s New
Since 2014 when CADD was first released there have been four score updates. In summary, the main modifications were changes to the machine learning algorithm and software library. For those tech savvy individuals, the predecessor, CADD v1.0, used a linear support vector machine whereas CADD v1.4 integrated a logistic regression model based on SciPy and scikit-learn. Beyond those changes, and on a more diagnostic perspective, CADD now provides scores for the human genome build GRCh38 (CADD v1.5) in addition to scores for GRCh37 (CADD v1.4). In total, CADD v1.4 covers 2,937,693,113 bases on GRCh38 compared to 2,858,658,094 bases on GRCh37 (Rentsch, 2019). For further differences between CADD v1.4 and CADD v1.5, please visit their direct content.
Utility in VarSeq
Like our other annotation sources, CADD scores are available in the Secure Annotations directory (Add>Variant Annotation) and are integrated in the VSClinical ACMG guidelines. Although the CADD annotation can be downloaded internally, the file is approximately 52GB, which is quite large. Fortunately, rather than downloading this large file it can be pulled from the Golden Helix server and be directly integrated into your project, without using your internal memory. The CADD annotation pulled from the server then computes scores for every variant in your project at a rate of roughly 30,000 variants per minute. In VSClinical, CADD scores are displayed in the Computation Evidence section alongside the in-silico predictions and will contribute to the ACMG criteria PP3, which states: Multiple lines of computation evidence support a deleterious effect on the gene or gene product.

Figure 3: CADD scores are precomputed for all variants in your project and are displayed in VSClinical
Conclusion
We are very excited to have the updated CADD scores for GRCh37 and GRCh38 now fully integrated into VarSeq and available for your clinical variant interpretation and reporting workflows. If you are a VarSeq user and would like to have this algorithm added to your package, just ask your area manager for a trial of the CADD add-on. If you are thinking about using VarSeq for your analysis for the first time, now is a better time then ever – request an evaluation here!
Citations
Rentzsch, Philipp et al. (2019) “ CADD: Predicting the Deleteriousness of Variants Throughout the Human Genome” Nucleic Acids Research, 2019. Vol. 47 doi: 10.1093/nar/gky1016