About Julia Love
Julia Love is a Genomic Curator and Product Quality Manager at Golden Helix, joining the team in June of 2019. Julia graduated from Kennesaw State University with a bachelor's degree in Biology, followed by earning a master's degree in Molecular Biology with a focus in Neuroscience from Boise State University. When Julia is not providing support and training to Golden Helix customers, Julia enjoys backpacking, canoeing, and spending time with friends and family.
With the release of VarSeq 2.2.4 just around the corner, I want to detail some new 2.2.4 features that will enhance somatic variant annotation and fusion analysis within VSClinical. A webcast back in July showed some of these updates in action, so if you are looking for some more content on this topic, I would highly recommend checking out the… Read more »
We had a wonderful turnout for our recent webcast, “Reduce Turn-Around with Enhanced Cancer Annotations & Golden Helix CancerKB Updates.” Thanks so much to those who were able to join us! And not to worry, we have a link to the recording here just in case you weren’t able to make it. We covered the gamut of new and updated annotation tracks available in VarSeq along… Read more »
VSPipeline is becoming a very popular tool among VarSeq users as it is essential for creating repeatable clinical workflows that can be executed in automated fashion. Since VSPipeline is a command-line tool, I think it would be helpful to discuss some of the best practices along with helpful tips for getting the most out of VSPipeline. Some of you may be less familiar with VSPipeline, so I want to cover how to set up the first run along with sharing the helpful tips as they arise. … Read more »
A common feature request from Golden Helix customers is to curate and make available genome assemblies for different plant and animal species. These requests commonly come from SVS users as many research projects are being carried out, and having the genome assembly available for analysis is essential. That being said, Golden Helix has an SVS Tutorial available that walks users… Read more »
Thanks for checking out this blog on how to to get more out of your VarSeq projects! The best way to show off one of my favorite new features that have been incorporated into the latest VarSeq 2.2.3 release is with a scenario: You work in a lab that processes and analyzes multiple whole exome samples for copy number variations,… Read more »
I want to take this opportunity to highlight and briefly discuss some of the key features and updates that have been incorporated into VarSeq 2.2.3. Some of you may have attended the webcast that covered the prominent new features added to VarSeq, which are the updates to improve whole-exome analysis workflows, namely improved CNV calling in whole-exome datasets. However, there… Read more »
Thank you to those who attended the recent webcast, “Exome Analysis with VS-CNV & VSClinical: Updated Strategies & Expanded Capabilities”. For those who could not attend but wish to watch, here is a link to the recording. In this webcast, we covered the capabilities and updates that have been incorporated into VarSeq that enhance whole exome sequencing workflows. The new… Read more »
In this blog, we will be covering new assessment catalogs and how they work to improve saving and tracking variant interpretations. VarSeq is a variant analysis tool that effectively analyzes single nucleotide (SNVs) and copy number variants (CNVs) in both cancer and germline workflows. Because VarSeq enables such diverse variant analysis, there are many research labs and institutions that evaluate… Read more »
Next-generation sequencing generates an immense amount of data which is then subject to a multi-step process to establish a validated bioinformatic pipeline. From processing raw sequence data to the detection of genetic mutations, establishing a validated and consistent bioinformatic pipeline makes a huge difference in the quality of patient care and accuracy of results. In this blog, we are focusing… Read more »
There is a multitude of interesting new features that have been incorporated into VarSeq 2.2.2. In this blog, I want to continue the discussion of these features and how each can be incorporated into your workflow, and also discuss the application of the Probability Segregation algorithm for copy number variation (CNV) analysis. The Probability Segregation algorithm is a new algorithm… Read more »
Annotating genomic variants is a very complex process but perhaps the most important part of next-generation sequencing variant analysis. Here at Golden Helix, we recognize the importance and value of having the most up-to-date sources available and curating new annotation sources as they become available for variant analysis. Golden Helix has curated over 100 annotation sources for human variant analysis… Read more »
VarSeq 2.2.2 was released on December 17th, 2020 and the main feature that was added to VarSeq was that the VSClinical ACMG Guidelines workflow now has an additional CNV interpretation framework based on the ACMG/ClinGen guidelines. This product supports interpreting CNVs detected with VS-CNV or imported CNVs alongside variants and requires both a VSClinical ACMG license and a CNV license…. Read more »
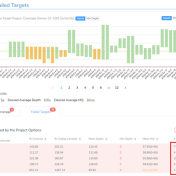
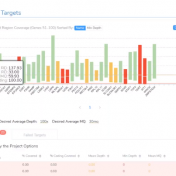
Curious about how coverage statistics can be used in conjunction with VarSeq? Evaluating the coverage over target regions or whole genomes is essential whether you are working with variant or CNV analysis. VarSeq has had the capability to compute sample level coverage statistics for some time now, but in the 2.2.2 release of VarSeq, there are some new features that… Read more »
Webcast Recap In the recent webcast “Exploring New Features and Clinical Reports in the ACMG Guideline Workflow”, Gabe and I took viewers through an evaluation with CNVs and SNVs according to the ACMG Guidelines where we generated and customized a clinical report. Along the way, we highlighted many new features that will soon be available in the upcoming VarSeq release…. Read more »
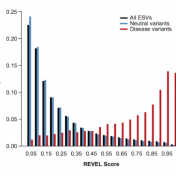
Typically, researchers are looking for rare variants in their next generation sequencing datasets. However, most of the nonsynonymous variants have unknown significance because there is an inherent difficulty in validating large numbers of rare variants or even detecting rare variants with high statistical power. In lieu of this issue, computational tools are needed as they accurately predict the pathogenicity of… Read more »
Did you know you can control your preferred transcript settings for clinical interpretation in VSClinical? Your lab is analyzing the DNA of a tissue sample from a patient with small cell lung cancer. The lab technician has imported the patient data into VSClinical to detect clinically relevant variants and evaluate and score these according to the AMP Guidelines, as well… Read more »
It is common knowledge that variants can be germline or somatic depending on whether the variant was inherited or acquired after birth. A well-known example is cancer-causing mutations in the BRCA genes, wherein the mutation may or may not have been inherited. Understanding the origin of the cancer-causing mutation is important when assessing potential treatment options as well as identifying… Read more »
SVS offers several options to conduct genome wide association tests and mixed linear models. At times, it can be challenging to decide which test, model, or adjustments to use when setting up your analysis. I want to briefly explore the options available in SVS for association tests, and mixed linear models to hopefully facilitate in understanding and choosing which options… Read more »
Thank you to everyone who joined me for yesterday’s webcast, I hope you all enjoyed it. If you missed the live event and are interested in knowing what we talked about, good news, you can watch the recorded version right here! There were so many great questions asked during our Live Q&A that I was unable to answer all of… Read more »
Golden Helix ships a variety of templates that are designed to provide a starting point for users to evaluate variants in VarSeq. Naturally, as users become more familiar with the software, there is a desire and necessity to tailor the template design to accommodate a more thorough variant analysis. To add to these template customizations there are several algorithms and… Read more »