Typically, researchers are looking for rare variants in their next generation sequencing datasets. However, most of the nonsynonymous variants have unknown significance because there is an inherent difficulty in validating large numbers of rare variants or even detecting rare variants with high statistical power. In lieu of this issue, computational tools are needed as they accurately predict the pathogenicity of rare variants and find the variants that are the most likely to cause disease. There are many tools available that can predict the pathogenicity of missense variants, however, these tools are not always in agreement as different features are used to determine the pathogenicity like amino acid, nucleotide conservation, or biochemical properties. These issues are exacerbated when they are applied to rare variants. Fortunately, the rare exome variant ensemble learner (REVEL) method offers a solution and is available to annotate variants within VarSeq!
REVEL has been around since October of 2016 and was trained using pathogenic and neutral rare missense variants. REVEL predicts the pathogenicity of missense variants based on a combination of scores from 13 individuals tools including MutPred, FATHMM v2.3, VEST 3.0, PolyPhen-2, SIFT, PROVEAN, MutationAssessor, MutationTaster, LRT, GERP++, SiPhy, phyloP, and phastCons. It is important to mention that when REVEL’s performance was compared to individual tools and seven ensemble methods, REVEL had the best performance overall and performed the best in distinguishing pathogenic from neutral variants (Figure 1). The REVEL method scores rare missense variants on a scale ranging from 0-1 with a score of 1 correlating with a greater likelihood of that variant being disease-causing.
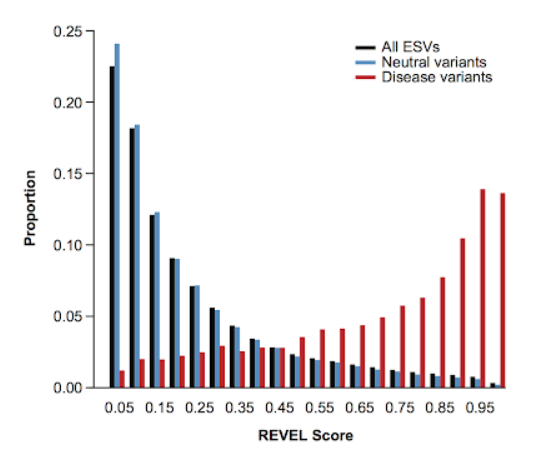
Figure 1: The REVEL score distributions for disease mutations (in red), neutral rare variants (light blue), and exome sequence variants (dark blue)
In VarSeq, REVEL is available as an annotation source that can be used for both GRCh37 and 38 based projects. To download and add REVEL to your project go to Add > Variant Annotation > Public Annotations. The output from the database can be used in workflows to filter variants to those that have higher REVEL scores. Figure 2 shows an example of the output from REVEL in VarSeq wherein I chose to filter down to variants that have REVEL scores of 0.7 or higher. In the example, the REVEL scores are shown side-by-side with output from RefSeq Genes and the ACMG Classifier algorithm. Together, the clinical impact of a variant can be evaluated utilizing the score output from REVEL and the variant classification according to the ACMG guidelines.
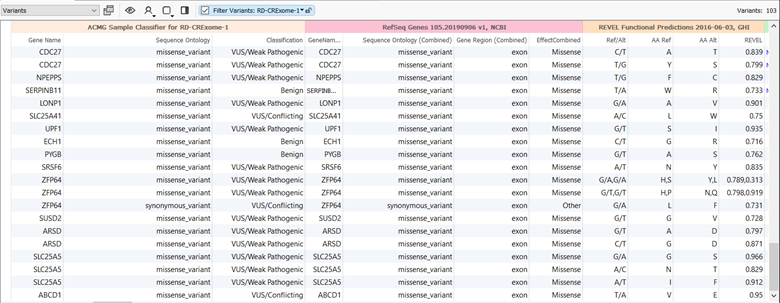
Figure 2: An example of the output from REVEL in VarSeq
I hope you enjoyed reading this blog and learning a little bit more about REVEL and how you can use this source in your VarSeq projects. For more annotation updates and workflow tips, feel free to take a look at our other blogs and webinars! Thank you for reading, and if you have any questions feel free to contact support@goldenhelix.com or, if you would like to request a free trial of our software you can contact info@goldenhelix.com.