Low read depth? Great!
We are excited to introduce our new CNV calling algorithm for low and ultra-low read depth Whole Genome Sequencing (WGS) data. This algorithm is designed to call large cytogenetic events with high confidence from low read depth whole genome data, with as few as one million aligned reads or 0.02x coverage. The low sequencing cost of these low read genomes, combined with the ease and efficiency of our algorithm, make it an intriguing alternative to existing methods used to identify large chromosomal abnormalities.
From BAMs to Karyotypes
The process of calling CNVs from WGS data begins by importing your BAM files into VarSeq and running our new Binned Coverage Statistics algorithm. This algorithm divides the genome into a number of equal-width bins and then computes coverage for each bin. This coverage data is then utilized by our CNV caller to detect cytogenetic events.
For ultra-low depth whole genomes, we suggest bin sizes of 1 Mbp (million base-pairs). The next step is to run the new CNV Caller on Binned Regions algorithm designed specifically for calling large cytogenic sized events with these bins.
As with our existing Target Region CNV Caller, event detection is performed by comparing the coverage of each bin to a set of reference samples. We then use our own CNAM optimal segmentation algorithm to segment the genome and identify CNVs.
Because our goal is to only detect large cytogenetic events, there is no need to rely on a complex probabilistic model. Our segmentation algorithm allows us to perform this task with exceptional speed and efficiency, often providing results within minutes.
In addition to calling these chromosomal abnormalities, we also provide complete karyotype notation for each event, describing the relevant chromosome along with the affected bands.
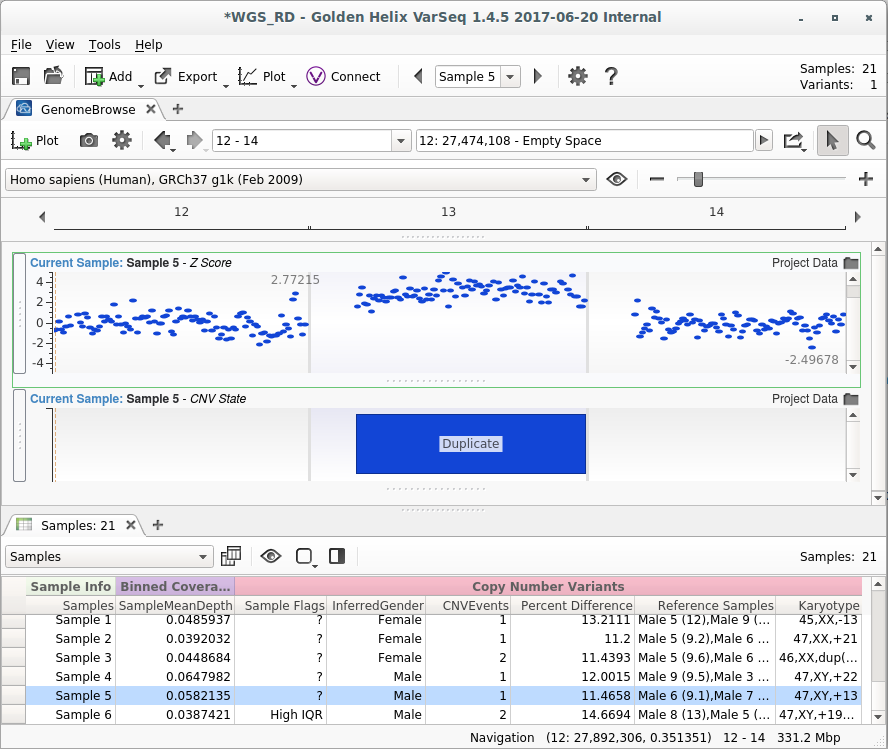
In the above example, a chromosome 13 aneuploidy was called with high confidence. The provided karyotype notation is “47,XY,+13”, denoting that this sample is a male with 47 chromosomes, and a duplication of chromosome 13. The Z-Score shows the number of standard deviations each bin is from the reference set. Because the bins in chromosome 13 are an average of three standard deviations from our references, we have strong evidence for this call.
Inexpensive and Easy Karyotyping
With our real-world test data, we have found you need as few as a million reads to get this level of accurate chromosomal aneuploidy calls, including sex-chromosome calls such as XXY.
Since sequencing cost is a function of the number of reads, and whole genomes require very simple sample library preparation with no target capture kit costs, this allows for very inexpensive sequencing and results in BAM files in the 50-100MB range!
We may see more and more labs adding ultra-low read depth assays to their mix alongside targeted gene panels to get both the targeted and the karyotype level picture with one sequencing machine.
Try It Out
If you are interested in trying out our WGS CNV caller or are interested in adding CNV to your existing VarSeq license, please reach out to info@goldenhelix.com. Our team of experts would be happy to demonstrate how you can use this powerful new feature.