Revisiting the Five Splice Site Algorithms used in Clinical Genetics
Interpretation of variants in accordance with the ACMG guidelines requires that variants near canonical splice boundaries be evaluated for their potential to disrupt gene splicing [1]. The five most common tools for splice site detection are NNSplice, MaxEntScan, GeneSplicer, HumanSplicingFinder, and SpliceSiteFinder-like. Because these algorithms have been made easily accessible in the bioinformatics tool Alamut, they have been canonized for the purpose of evaluating a variant’s impact on gene splicing in clinical genomics. While these methods are often treated as black-box oracles on equal footing when used to classify variants, the results presented here show that the performance of these methods varies wildly.
Five Methods, Four Approaches
Many splice-site prediction methods, such as HumanSplicingFinder and SpliceSiteFinder-like, trace their origins back to the work of Shapiro and Senapathy in 1987 [2] [3]. In this landmark publication, the authors describe a splice site detection method based on position weight matrices (PWMs). The PWMs used by Shapiro and Senapathy encode the relative frequencies of each nucleotide for a specified window surrounding a splice site. While the size of the windows and equations used to compute scores varies between methods, our results indicate that both PWM-based algorithms achieve similar performance.
In 1997, Reese et al. investigated the use of neural networks for splice site prediction with their algorithm, NNSplice [4]. This approach converts a genomic sequence into a binary string, where each pair of nucleotides is represented by 16 bits. These binary strings are used as input to a neural network with a single hidden layer and a single output neuron. The donor network has 15 inputs and 2 hidden units, while the acceptor network has 41 inputs and 10 hidden units. Both networks are trained using backpropagation.
In 2004, MaxEntScan was developed by Yeo et al [5]. This method models splice site sequences using the Maximum Entropy Distribution given a set of constraints defined as low-order marginal distributions. A greedy search algorithm is used to select the constraints used by the model. One benefit of this method is that it provides a relatively unbiased approximation of the distribution of sequence motifs, assuming very little about the underlying data.
GeneSplicer, developed in 2001 by Pertea et al. uses maximal dependence decomposition in conjunction with Markov models for splice site detection [6]. The goal of maximal dependence decomposition is to identify the most significant dependencies between positions associated with a splice site motif. The algorithm learns Markov models for the total sequence, the coding region, and the non-coding region surrounding splice sites, incorporating the identified dependencies. These models are then combined to score potential splice sites.
To implement these algorithms, we relied on the published papers and in the cases where source code was provided, followed the algorithmic strategy faithfully. The PWM methods have no source code provided, but the Shapiro and Senapathy included the weight matrices used in the publication. Naturally, any component of these algorithms not included in the publication, or developed outside of the academic literature cannot be faithfully reproduced. For example, you can go to the Human Splicing Finder website and run splice site predictions. This method has been made proprietary and will have differing results, but in our testing, the results were similar in performance. (We will refer to our implementation below as HumanSpliceFinder-like)
Benchmarking on The Human Spliceosome
We compared these algorithms on both donor and acceptor splice site data in terms of accuracy, sensitivity, specificity, and precision. For these experiments, we extracted 20,000 known splice sites from the GRCh38 human reference sequence using exon boundaries specified by NCBI RefSeq genes. This data was combined with 20,000 false splice sites from the HS3D splice site dataset. The performance of the algorithms on this data is presented in Tables 1 and 2 below.
Table 1: Donor Splice Site Results
| Accuracy | Sensitivity | Specificity | Precision | |
| HumanSpliceFinder-like | 78.4 | 82.2 | 78.0 | 26.6 |
| SpliceSiteFinder-like | 79.7 | 82.4 | 79.5 | 28.0 |
| MaxEntScan | 87.0 | 72.9 | 94.9 | 88.9 |
| NNSplice | 87.1 | 83.6 | 94.4 | 96.9 |
| GeneSplicer | 88.2 | 85.4 | 93.8 | 96.6 |
Table 2: Acceptor Splice Site Results
| Accuracy | Sensitivity | Specificity | Precision | |
| HumanSpliceFinder-like | 85.6 | 85.4 | 85.9 | 92.4 |
| SpliceSiteFinder-like | 82.7 | 81.8 | 84.4 | 91.3 |
| MaxEntScan | 89.5 | 89.0 | 90.3 | 95.1 |
| NNSplice | 81.5 | 77.4 | 90.0 | 94.2 |
| GeneSplicer | 92.0 | 92.9 | 90.1 | 95.1 |
These results show that GeneSplicer provides the best overall performance, achieving high accuracy, sensitivity and precision for both donor and acceptor sites. Although NNSPLICE and MaxEntScan had lower sensitivity, when compared to GeneSplicer, these methods also achieved high accuracy and specificity. Overall these two methods are comparable, with NNSPLICE outperforming MaxEntScan on donor regions, but under-performing on acceptor regions. The PWM-based methods, HumanSpliceFinder-like and SpliceSiteFinder-like, performed well on acceptor regions but had the worst overall performance on donor regions due to an extremely high false positive rate.
Splice Site Prediction as Part of the VSClinical
We will soon be releasing VSClinical, a product that enables the interpretation of variants following the ACMG Guidelines. Through standardized questions, automatically computed evidence, and historical context, VSClinical provides clinicians with everything they need to classify variants consistently and reproducibly, thereby reducing the subjectivity of the variant classification process.
VSClinical has robust support for splice site prediction, including the ability to predict the effect of each variant on its nearest splice site, with associated scores from four of the five algorithms discussed above. We chose to include only one of the PWM-based algorithms due to their similar strategy and performance, opting to include the method described by Shapiro and Senapathy. For each of these algorithms, we provide a 0 to 1 score, indicating the confidence of each algorithm on the presence of the nearest splice site, along with a prediction of whether the algorithm predicts the variant to be disruptive. Additionally, we provide a count of the total number of algorithms predicting the variant to be disruptive. This information is presented via an intuitive user-friendly display and is used as evidence to automatically suggest answers to specific ACMG criteria.
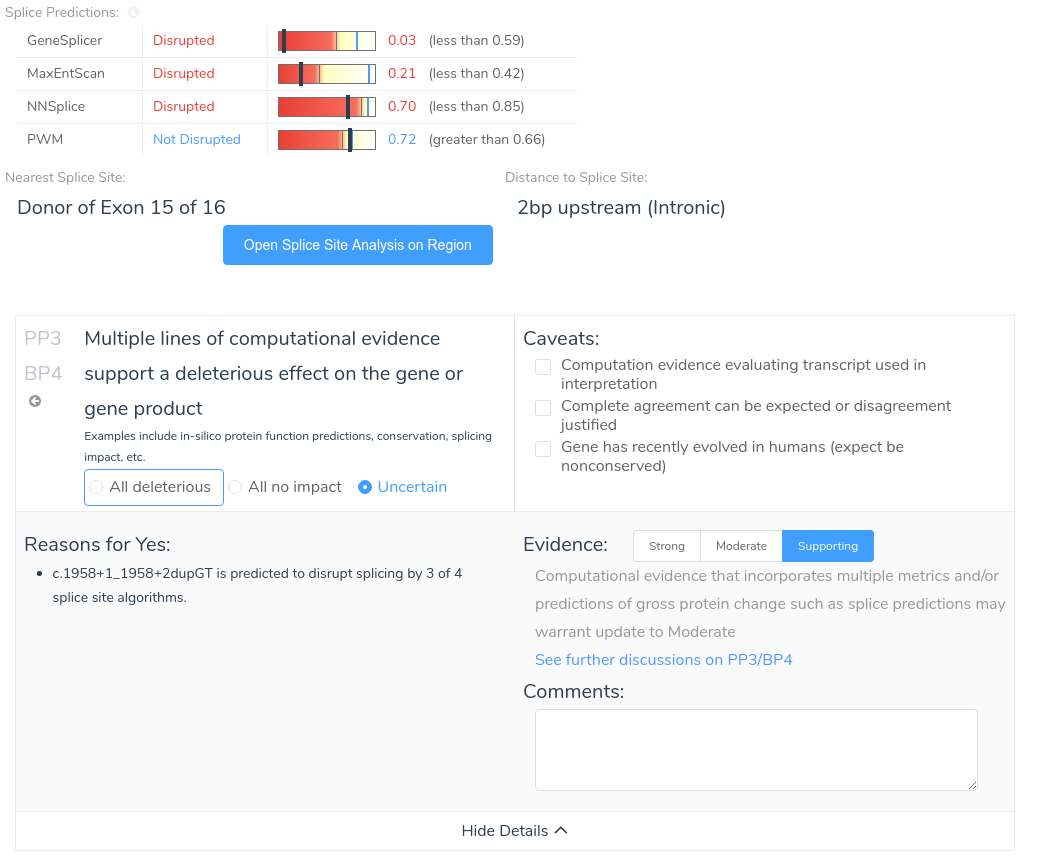
VSClinical also provides an intuitive visualization of each variant’s effect on splicing as predicted by each algorithm. This allows users to easily identify the introduction of novel splice sites, as well as disruptions of existing splice sites.

We look forward to providing this functionality and much more in our upcoming VarSeq release next month!
References
| [1] | S. Richards, N. Aziz, S. Bale, D. Bick, S. Das, J. Gastier-Foster, W. W. Grody, M. Hegde, E. Lyon and E. Spector, “Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology,” Genetics in medicine, vol. 17, no. 5, pp. 405-423, 2015. |
| [2] | F.-O. Desmet, D. Hamroun, M. Lalande, G. Collod-Beroud, M. Claustres and C. Beroud, “Human Splicing Finder: an online bioinformatics tool to predict splicing signals,” Nucleic acids research, pp. e67–e67, 2009. |
| [3] | M. B. Shapiro and P. Senapathy, “RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression,” Nucleic acids research, vol. 15, no. 17, pp. 7155-7174, 1987. |
| [4] | M. G. Reese, F. H. Eeckman, D. Kulp and D. Haussler, “Improved splice site detection in Genie,” Journal of computational biology, vol. 4, no. 3, pp. 311-323, 1997. |
| [5] | G. Yeo and C. B. Burge, “Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals,” Journal of computational biology, vol. 11, no. 2-3, pp. 377-394, 2004. |
| [6] | M. Pertea, X. Lin and S. L. Salzberg, “GeneSplicer: a new computational method for splice site prediction,” Nucleic acids research, vol. 29, no. 5, pp. 1185-1190, 2001. |