Scaling is in our DNA: Making Genomics Accessible
One of the things I absolutely love about the work we do at Golden Helix is keeping up with the changes in data analysis driven by the iterative and generational leaps in technology.
But one thing has always been a constant since day one: we break preconceived notions of what scale of data a user can wrangle and analyze on their own single machine.
As we saw the density of microarrays go from tens of thousands of probes to millions, with studies similarly scaling in the number of samples, we carefully constructed our data structures and algorithms to swap in and out pieces of these large matrixes to display spreadsheets, run statistics and plot the per-marker results (in the iconic “Manhattan Plot” for example).
In fact, we are still tweaking our SNP and Variation Suite in this regard. In our last release we found room for more optimizations to improve Manhattan Plot render speed (and other numeric dot plots).
And as incredibly as this would have seemed 5 years ago, we are proud to be supporting customers who have used SVS to tackle their studies with the number of samples (N) in the 100,000+ range!
We are also working closely with some of our animal breeding customers to optimize the more advanced matrix-math heavy mixed model association techniques like EMMAX to remove the limits incurred by the (N x N) sized allocations required of these operations. While not a simple optimization (we have to build upon specialized mathematical techniques with domain-specific requirements in very novel ways), enabling our users to perform analysis on hitherto inaccessibly large datasets makes it worth it. Stay tuned as we announce the release of these “Large N” scaled algorithms!
All of battle-hardened engineering tricks to build fast, scalable core data structures and algorithms came to bear on on the task of building VarSeq: A desktop based tool that wrangles whole genomes with instantaneous feedback on the interactive act of adding and adjusting filters, visualizing variants and running multiple annotation and genomic algorithms concurrently.
As that same core technology now powers our recently revealed VSWarehouse: a genomic data warehouse for NGS variants, we are continuing to invest in the engineering work to raise the scaling bar both in small incremental optimizations and big projects.
One of the big projects I’m very excited about is the from-the-ground-up construction of a new memory-optimized table grid view for VarSeq.
The file format and algorithms in VarSeq happily scale to tens of millions and even hundreds of millions of variants. In other words, the full expected range of NGS data, given that the 2,500 whole genomes sequenced by the 1000 genomes Phase 3 project resulted in 85 million unique variants.
But in practical terms, VarSeq has had a displayable row limit of far less (in the 15-20 million range) due to the table technology we had been building on that required some memory be allocated per-row.
Along with a list of advanced features and usability improvements we have been storing up, the new from-scratch table grid we are building is designed to have zero overhead per-row and thus dramatically improve the speed in which a whole genome NGS project can be opened and interacted with while eliminating the bounds to table size.
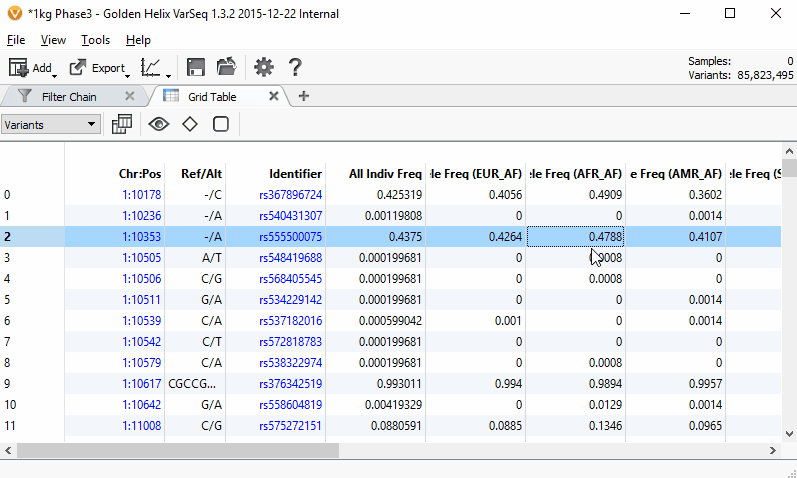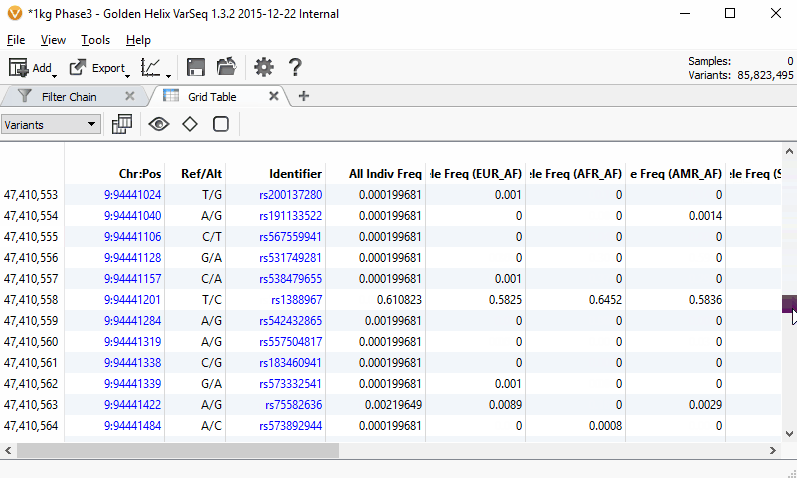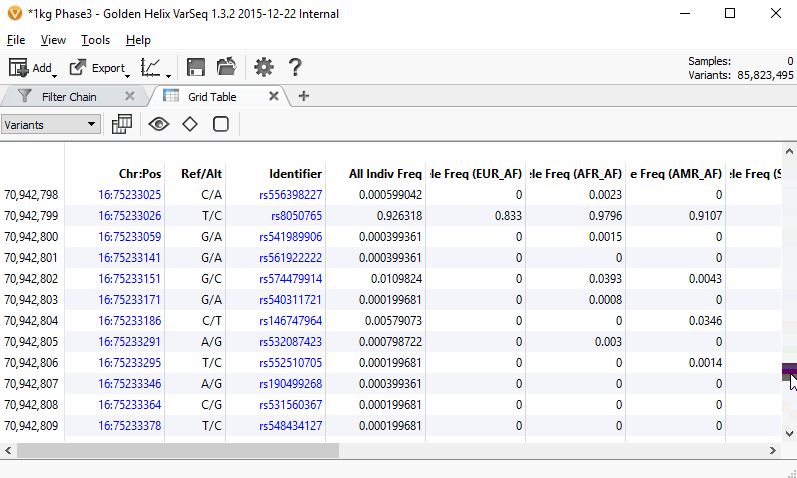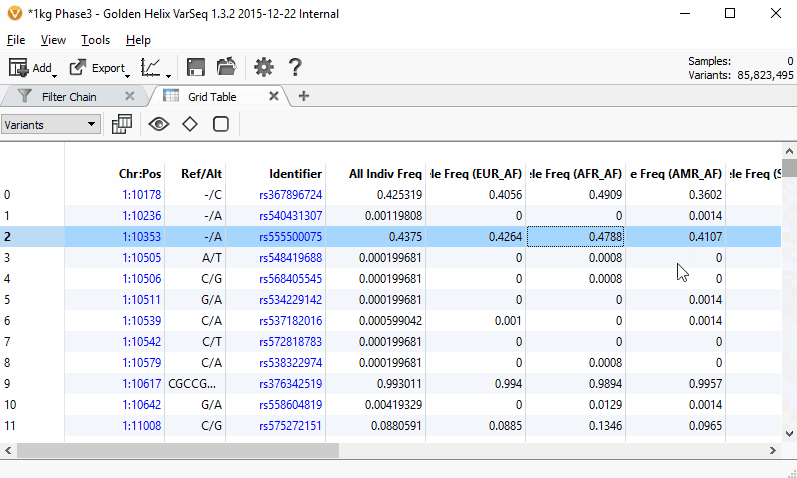
Scrolling through the 85 million variants from the 1000 Genomes Phase3 project while only taking ~70MB of RAM with the new VarSeq table grid.
While most users may only notice some of the cosmetic and added features of the new grid, we are happy to stay ahead of the technology and adoption curve while removing limits of large-N whole-genome analysis.
Whole genomes are becoming both economical and in many ways a “better exome” through its simplified prep, uniform and unbiased coverage, and ability to assay copy number.
Overall I expect to see the usage of whole genome sequencing grow dramatically, and with it the interest in analyzing and warehousing cohorts of WGS samples.
Golden Helix is committed to continuing its leadership in making genomic analysis accessible to individuals while scaling to the needs of researchers, clinical labs, institutions and consortiums.
Glad to be one of your customers ‘egging’ you on to help deal with the large N problem in Agriculture. Looking forward to your results.
That’s right Matt!
We are looking forward to you trying them out. Great progress being made.