Yesterday, it was my pleasure to share in a live webcast our integrated solution for genetic data warehousing, VSWarehouse.
If you missed the webcast live, feel free to check out the recording.
Although we had a great set of questions at the end of our presentation, we didn’t have time to answer all of them, so here is a selection of the remaining representative questions and my answers.
- Can you talk about system requirements for VSWarehouse? Does it run in the cloud?
We can provide a hosted version of VSWarehouse for you or it can be installed and hosted on your own local servers or on your own cloud. Additionally, VSWarehouse does not require big distributed clusters. It can be run on a single server, with modest system requirements and scale to thousands of exomes and will be no problem for storing thousands of targeted panel samples. A standard 4-8 core server with 16GB of RAM should be more than adequate.
- Can I run VSWarehouse locally and add samples to it directly from secondary pipelines? Will it still be able to leverage all the public and private annotation sources?
Yes, VSWarehouse is designed for locally managed installs. Although we demonstrated the ease of adding sample’s call sets from within a VarSeq project in the webcast, we will also have scripts and hooks to import VCF files directly into the server.This should allow a secondary analysis pipeline to warehouse the sample’s called variants as part of its automated process. All of VarSeq’s algorithms and annotations are still used in this model.
- How does the early Adopter Program work for existing clients?
If you are an existing customer, with current licenses, we will take a look at the existing equity in your current license and come to an amicable solution in order to provide you with access to VSWarehouse.
- Can you describe the capabilities of your API to connect with external systems?
VSWarehouse allows you to control access to the APIs as well as permissions down to a project level, allowing you to control who has access to certain types of samples. For example, you may want to limit access to research samples versus clinical samples.The APIs provide a SQL view as well as a higher level REST API and local python data model access that is designed for building and running scripts that will give you alerts when things change and allow you to run back-end integration, etc.
- Can I use this to share data with other researchers?
Absolutely. With VSWarehouse, licensed VarSeq users can contribute add and update data to individual projects, clinical reports and assessment catalogs. However, you can also choose to grant read-only access to any number of people to view and query the data in the system.
- What annotation tracks are included in VSWarehouse?
Golden Helix dedicates time to curating various annotation tracks, both public and commercially licensed. You are also able to easily create annotation sources your own internal catalogs or cohorts available through dbGaP or other controlled access consortiums. VSWarehouse can pull in any of the annotations already available in VarSeq such as ClinVar, OMIM, dbSNP, COSMIC, ExAC 60K exome frequencies, SIFT and PolyPhen to name a few.
- Is VSWarehouse limited to human genomes? What about the new GRCh38 human reference?
Built on the core VarSeq technology, VSWarehouse’s individual projects have an associated genome reference and can certainly be set up for the new GRCh38 human reference as well as other organisims with reference genomes such as mm10 for mouse.We have quite a few annotations for GRCh38 in our public annotation repository, including NHLBI 6500, ClinVar, and dbNSFP Functional Predictions.
- How does it handle genes with multiple isoforms/transcripts where the impact of a variant is different for the different isoforms?
One of the strengths of VSWarehouse is how it leverages on the core annotation algorithms we have developed for VarSeq. For gene annotations, we provide per-transcript Sequence Ontology and descriptive attributes for variants. We then aggregate these into a “Combined”, worst effect fields at the gene level. Finally, we use public annotations and heuristics to define the Clinically Relevant Transcript from the multiple isoforms and provide its key attributes at the gene level.
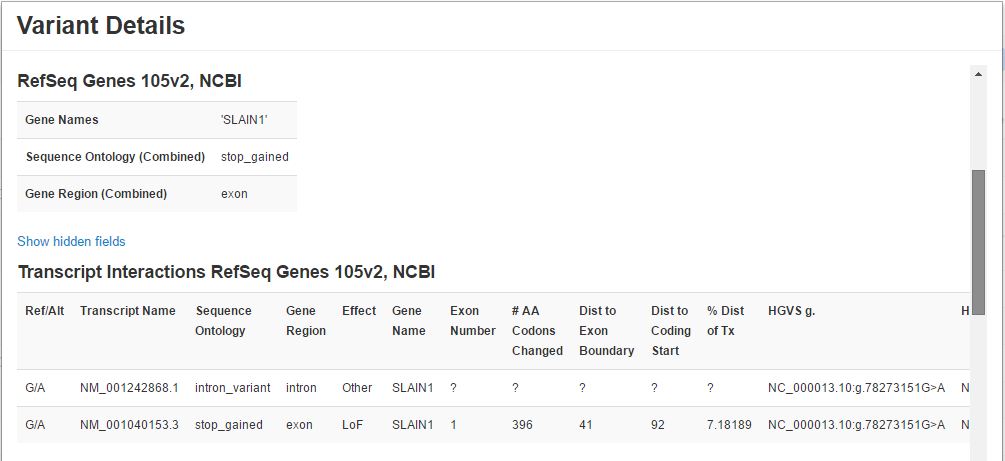
Example of viewing the variant details in VSWarehouse of a mutation with differing sequence ontologies in the two transcripts of SLAIN1. The “Combined” Sequence Ontology at the gene level is the ranked most-damaging classification of “stop_gained”.
- You demonstrated using VSWarehouse to store the sample variant calls and reports for trios (a patient and their parents). Can analysis be done on more extended lineages?
VarSeq is being used in both the clinical and research setting to find candidate causal variants in disease in complex pedigrees, often with dominant inheritance model suspected as well as trios, quads and singletons. While the analysis in VarSeq is focused on a single affected individual at time, you can filter on shared presence or absence of variants of any relations’ variants. For example, you can filter to variants shared in a co-affected sibling, but not shared by an unaffected uncle etc. See my blog post on Handling Singletons & Complex Pedgrees with our Gene Count Algorithms for more examples. VSWarehouse will happily store all the samples, and can compute allele counts and frequencies for different “sub-cohorts” such as your Affected vs Unaffected.