There is no doubt that we have big data in the field of genomics in general and Next Generation Sequencing specifically.
Illumina’s latest HiSeq X can produce 16 genomes per run, resulting in terabytes of raw data to crunch through.
Yet all that crunching is not the hard part.
So, what is the main obstacle to scientists being able to handle genomic big data?
In short: variant analysis is not a solved problem and in fact we are still working out basic standards and methods for grading and bench-marking our tools.
Bioinformatics algorithms, reference data sets and “best-practice” tools are fast-changing and often driven by researchers, not commercially supported software engineers.
We are just getting around to having the community discussions, such as those in the GA4GH working groups, on representing things like variants and their annotations, at a fundamental data level.
The process of aligning and calling variants is embarrassingly parallel, and bundled solutions from Illumina and Ion Torrent generally meet the “good enough” threshold. This is especially true for targeted gene panels, which accounts for the majority of sequencing in the clinically market.
At a maximum, a sequenced whole genome may result in nearly a billion short reads aligned to the genome and stored in a 300GB BAM file. The roughly 10 million variants called against the reference is stored in a less than 1GB compressed gVCF file.
But at the end of the day, there will only be around 50,000 variants in genes and even fully annotated, those takes up just a few megabytes.
After this point, working on a single sample, we are not doing analysis on “big data”. In fact, it can very comfortably be done on any desktop or laptop computer.
But, even though the analysis is now in the small, it is far more complex, integrative and requires human judgment.
This is where Golden Helix focuses its efforts: providing commercially supported variant annotation, interpretation and reporting solutions.
So are there roadblocks to big data analysis, and how can we address them?
The standard exome or genome “align and call” pipelines have become what most informatics infrastructure should become: boring and predictable.
Cloud platforms like DNAnexus provide an important service of putting this infrastructure into more individual’s hands. Along with the accessibility of desktop sequencers, the creating of NGS data and the running of secondary analysis should not be the roadblock.
Where the complexity kicks in is in the very interesting analytics that are closer to the outcomes. Successfully analyzed NGS data can lead to gene discovery and enables precision medicine by informing clinical care.
Variant annotation and interpretation integrates a constantly moving target of public genomic databases, a single sample of interest and if possible, the aggregate information of all previously assessed clinical samples.
This aggregate of samples and their variants (variant warehouse), has grown in value as a lab ramps up, is a critical component to creating the testing outcomes.
In aggregate, such a warehouse can quickly contain billions of genotype calls and all their accompanying metrics in dozens of fields (since the number of records = number of samples X number of variants).
Now we are back into big data territory, where even traditional transactional relational databases creak and bow without moving to complex distributed multi-machine systems.
To prevent this from becoming a roadblock, we have developed some specialized technology to make variant warehousing scale on a single server. Our compressed column-store is both storage efficient and wicked fast for doing analytical queries with a user-friendly UI or traditional SQL.
With genomics-aware representation of data, this storage technique is more efficient than compressed VCF files and optimizes genomic queries like:
- “How often have I seen this variant?”
- “Alert me when ClinVar changes classification of a variant I have reported on”
- “How many rare functional variants have I seen in this recently published gene?”
- “Have I put this variant into a clinical report for any previous samples?”
Let’s Stop Saying “Big Data”, But Talk about Its Parts
I had a hunch that the term “Big Data” was finally cooling off in 2015, and a Google Trend search seems to confirm that.
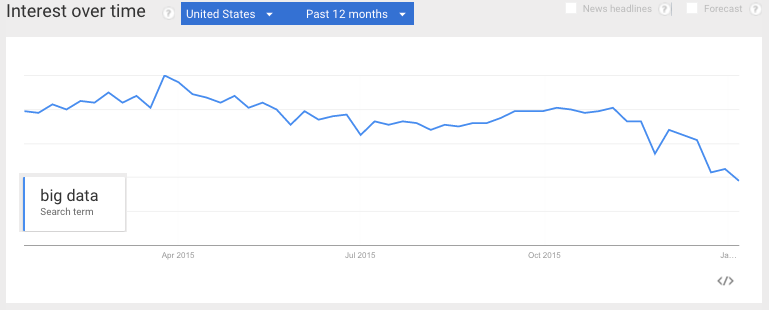
This article on Big Data Trends for 2016 points out a potential explanation:
Database management and data science technically fall under the same category at the moment, when the reality is that they are different.
So instead of talking about Big Data, lets talk more about more tangible challenges of data management and data analytics.
Stay tuned as we begin tackling the big data management problem of aggregating sequenced NGS samples and their variants. We will also address the big data analytics problem of making meaningful subset queries, aggregates and cross-references to public and private genomic databases and annotations.