We have a lot to thank the 1000 Genomes project for in the genomics community. By the collaborate efforts of many researchers and organizations, the project produced not only the first catalog of rare human variation but in the process standardized many things we take for granted, such as the VCF and BAM file formats.
The variant frequencies of the 2500 individuals in the final Phase 3 of the project is what we first think of now when referencing the “1KG” dataset. These variant calls were refined as the capstone of the entire project, bioinformatically welding together genomes, exomes, genotype arrays and some esoteric other datasets to create a single high-quality and heavily polished set of small variants and CNV deletions.
There are a couple good reasons to still use the 1000 genomes variant frequencies, even in the era of the population catalogs like gnomAD that far exceed its sample count, and so I spent some time recently producing a new version of it using VarSeq that will make it even more useful for the clinical interpretation of rare diseases.
The Healthy Person Filter
There are a number of times in the variant interpretation process where you want to know if a variant is present in healthy adults. While there is no public database that can guarantee every individual in it is healthy, the ESP6500 dataset is actually the opposite. First released in 2011 by the National Heart Lung and Blood Institute, it was the first project to exceed 1000 Genomes in sample count and was produced by a consortium of researchers studying adult-onset diseases (as the institute name). In fact, many of the samples that were selected for sequencing had the most severe phenotypes for the purposes of discovering associations between their sequenced variants and the metabolic markers and other disease-specific phenotypes of the patients in the consortium.
In their footsteps, the ExAC project pulled together 60,706 unrelated individuals for exome sequencing from many research cohorts. As you can see at the bottom of the ExAC FAQ page, many of these samples are also from potentially diseased individuals, including 4K from ESP6500, 9K from T2D-GENES, 12K from the Swedish Schizophrenia & Bipolar Studies cohort. While they took care to remove individuals with known pediatric disorders, and these complex diseases are unlikely to skew population allele frequencies for the vast majority of variants, you may want to be cautious when ruling out a variant because it occurred in a small number of individuals from one of these databases.
In particular, the ACMG guidelines have some rules such as BS2 to be used as strong evidence that a variant is benign:
BS2: Observed in a healthy adult individual for a recessive (homozygous), dominant (heterozygous), or X-linked (hemizygous) disorder, with full penetrance expected at an early age.
For this rule to be scored as applied to a variant of interest, we need the variant to be homozygous in just one healthy individual!
But can we in good faith use ESP6500, ExAC or even gnomAD (superset of ExAC) to answer that question? I would argue no, and that leaves only one public population catalog that meets the threshold for “healthy adults”: The 1000 Genomes Project.
High-Quality Variants Now with Genotypes
Another outcome of the 1000 genomes bioinformatic efforts was a merging of multiple lines of evidence into their variant calls. While ExAC and gnomAD provide variant calls from GATK HaplotypeCaller with some additional QC and region flags added, they require the user to be aware of these potentially dubious QC-flagged variants. I previously wrote about an example of the type of false-positive variants that are present in ExAC (and gnomAD).
In contrast, the 1000 Genomes are already filtered and don’t include variants that have evidence of being in difficult to call regions. So in general, you are able to directly use it as a source of filtering out common and benign variants.
To support the type of analysis that requires not only variant frequency, but the awareness of the presence of a variant in a homozygous state, we need our population frequency annotation sources to have per-genotype counts. This granularity is available from ExAC and gnomAD, but unfortunately, the provided “sites” file with the equivalent data provided by the 1000 genomes project only contains “Allele Frequency”.
Thankfully, given the public nature of the genomes, the project also provides the raw genotype data for all the 2500 samples in VCF files! We recently imported the full set of these 85 million variants into VarSeq and used the integrated “Allele Counts” algorithm to produce our own Allele Frequency data to be exported as the new annotation track. And yes, VarSeq scales to operating at the scale of 200 billion genotypes no problem! So now, along with Allele Frequency, we also have Homozygous counts, Heterozygous counts and Allele Count in aggregate, as well as broken down by “super population”.
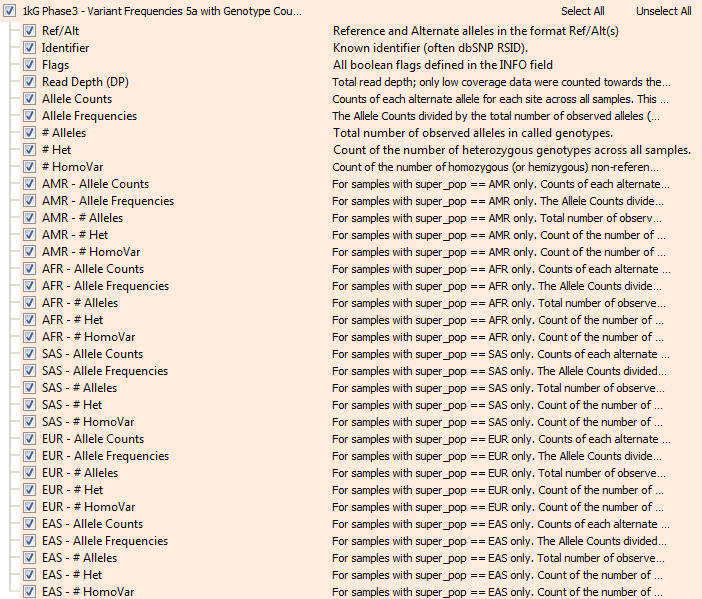
The new 1000 genomes variant frequency track in VarSeq with genotype counts.
Now from VarSeq, following the BS1 rule from the ACMG guidelines is as simple as adding “1kG Phase3 – Variant Frequencies 5a with Genotype Counts, GHI” as a variant annotation source, right-clicking on the “# HomoVar” field and choosing “Add to Filter Chain” and then configuring a filter like the one below that removes any variant that is Homozygous in one or more samples:
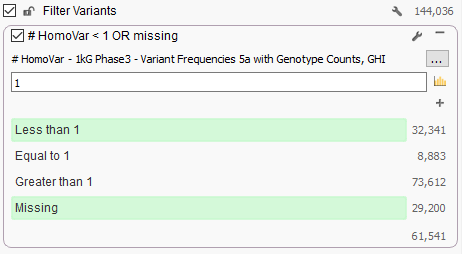
We spent a lot of time QCing this track, ensuring it matches the variant calls exactly as provided by the original frequency data produced by the consortium and we are pleased to make it available to our Golden Helix users. Check it out!